
Post-Exertional Malaise (PEM) and Remission: A Flare-Aware, State-Dependent Systems Model for ME/CFS and Long COVID
- Apr 24
- 67 min read
Updated: May 7
Reframing PEM as a multi-domain, threshold-dependent system failure and a core barrier to flare-aware, state-dependent remission in infection-associated chronic conditions
Part of the CYNAERA ME/CFS Library, a growing resource impacting post-exertional malaise, pacing, and remission for myalgic encephalomyelitis/chronic fatigue syndrome.
1. Executive Summary
Post-exertional malaise, or PEM, is the defining clinical feature of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and one of the most consistently misunderstood phenomena in chronic illness medicine. Although widely recognized in modern diagnostic frameworks, PEM is still frequently flattened into generic fatigue, deconditioning, or exercise intolerance, obscuring its true biological significance and contributing to diagnostic delay, therapeutic misinterpretation, and repeated failure in research design. PEM is not only the defining clinical feature of ME/CFS, but one of the most sensitive real-world indicators of whether the system is stabilizing or progressing toward remission (Institute of Medicine, 2015; NICE, 2021; Komaroff and Bateman, 2021; Stussman et al., 2020).
PEM is not ordinary tiredness after activity. It is a delayed, disproportionate, and multisystem worsening triggered by physical, cognitive, sensory, orthostatic, emotional, or environmental stress. In many patients, symptoms intensify hours to days after the initiating trigger and may persist for days, weeks, or longer. This pattern reflects a breakdown in recovery mechanisms rather than simple energy expenditure, with mounting evidence pointing to dysregulation across immune signaling, metabolic function, autonomic control, and neurovascular stability (Carruthers et al., 2011; Davenport et al., 2019; VanNess et al., 2010; Chu et al., 2020; Komaroff and Lipkin, 2021).
This paper argues that PEM should be understood as a threshold-dependent systems failure of adaptive capacity rather than a symptom of fatigue. The inability of conventional medical frameworks to capture delayed, nonlinear collapse has led to systematic underrecognition of severity, inappropriate management strategies, and poor endpoint selection in ME/CFS and related post-infectious illness research. When collapse is time-shifted, cumulative, and multi-domain, single timepoint measurement fails by design (Institute of Medicine, 2015; Natelson et al., 2019; Vink and Vink-Niese, 2020).
At a systems level, PEM reflects the interaction between cumulative load and a sensitized physiologic threshold. Total system burden is not limited to physical exertion, but includes cognitive demand, sensory input, autonomic strain, environmental exposure, and emotional load. When these domains are not jointly accounted for, patients exceed threshold through accumulation rather than obvious overexertion, producing the characteristic pattern of delayed crashes and incomplete recovery (Cook et al., 2017; Nijs et al., 2012; Rowe et al., 2014).
By centering PEM rather than treating it as one symptom among many, clinicians and researchers gain a more accurate framework for diagnosis, monitoring, subgrouping, and intervention design. In this view, PEM is not secondary to ME/CFS. It is the most clinically visible expression of the underlying systems instability that defines the illness and shapes its trajectory over time (NICE, 2021; Komaroff, 2019; Wirth and Scheibenbogen, 2021).
This framework also extends the state-dependent logic introduced in the abstract “CRISPR Remission: A Flare-Aware Gene Editing Pathway Engine for Immune-Volatile Chronic Disease,” presented at CRISPRMED26. While that work established a model for flare-aware therapeutic deployment, it did not fully articulate one of the most critical constraints governing intervention success in ME/CFS and related conditions: PEM itself. As a threshold-driven, delayed-response system behavior, PEM determines when an intervention is introduced into a stable versus unstable physiologic state. The PEM systems model developed in this paper provides the missing layer required to operationalize remission and intervention timing in complex chronic disease. By defining how cumulative load, threshold sensitivity, and delayed flare penalties shape system behavior, it enables more accurate identification of stability windows, more precise timing of intervention, and more reliable interpretation of therapeutic response. In this integrated view, PEM is not simply managed. It becomes a governing variable in determining whether recovery can be achieved and sustained.
2. What PEM Actually Is: Beyond Fatigue Models
From a modeling perspective, PEM can be understood as a threshold-dependent phenomenon governed by cumulative load across domains. Total system burden is not limited to physical exertion, but includes cognitive demand, sensory input, autonomic strain, environmental exposure, and emotional load. When these domains are considered together, the apparent unpredictability of PEM becomes structured and interpretable. Patients are not exceeding threshold because of a single identifiable event, but because multiple sub-threshold demands accumulate in ways that are not captured by traditional models of exertion. This interpretation is supported by evidence demonstrating that cognitive effort, orthostatic stress, and sensory load can independently provoke symptom exacerbation and physiologic strain in ME/CFS and related conditions (Cook et al., 2017; Nijs et al., 2012; Rowe et al., 2014; Davenport et al., 2019).
What emerges from both clinical observation and physiologic data is a consistent pattern. PEM behaves less like fatigue and more like a system failure signal. The features of that signal are remarkably stable across patients, even when the specific triggers differ, reflecting shared underlying mechanisms rather than idiosyncratic symptom expression (Institute of Medicine, 2015; NICE, 2021; Komaroff and Bateman, 2021).
Core Characteristics of Post-Exertional Malaise (PEM)
Multi-domain trigger sensitivity
Disproportionate response to demand
Delayed onset (temporal dissociation)
Prolonged and incomplete recovery
Threshold-dependent behavior
Multisystem expression
Hidden load accumulation
Variability and instability over time
Failure of adaptive recovery mechanisms
These characteristics are not independent observations. They describe a single underlying structure in which the body is operating with reduced tolerance to load and impaired ability to recover once that tolerance is exceeded. The combination of delayed onset and multisystem involvement is particularly important, because it explains why PEM is so often misinterpreted in both clinical and research settings. When cause and effect are separated in time, the connection between activity and outcome is easily missed, and the resulting instability may be attributed to inconsistency rather than physiology (VanNess et al., 2010; Snell et al., 2013; Davenport et al., 2019). To make this structure more explicit, PEM can be represented conceptually as a function of total system load relative to a dynamic threshold. This threshold is not fixed. It shifts based on baseline stability, prior exertion, environmental conditions, and recovery efficiency, consistent with evidence showing fluctuating physiologic capacity in ME/CFS across metabolic, autonomic, and circulatory domains (Fluge et al., 2016; Tomas et al., 2017; Systrom et al., 2021; van Campen et al., 2020).
In its simplest form:
Load_total = Lp + Lc + Ls + La + Le + Lm
PEM occurs when:
Load_total > Threshold(t)
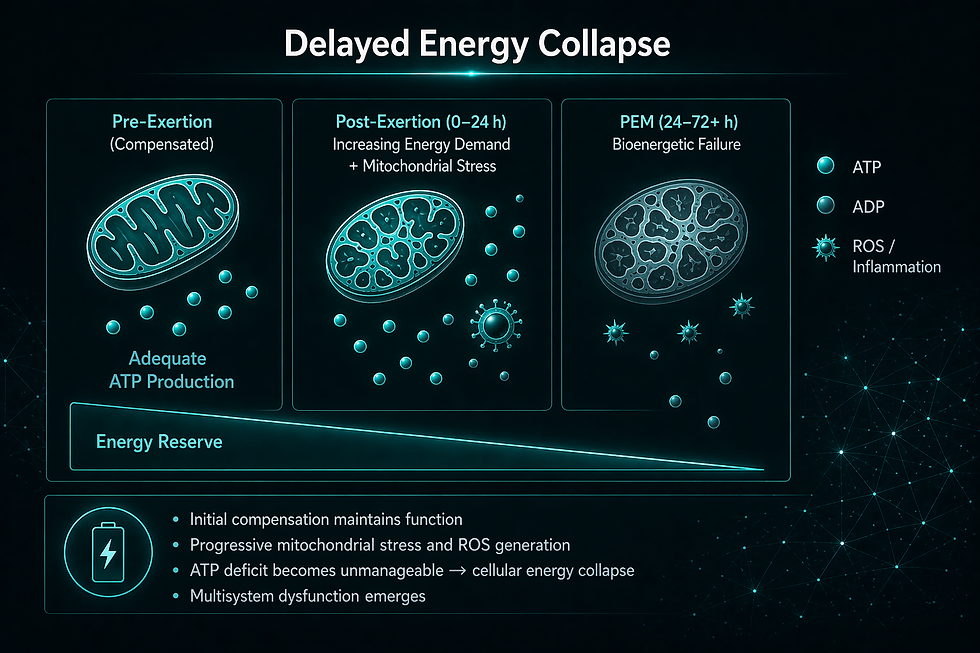
This framing makes visible what is otherwise hidden in standard clinical models. When only physical exertion is measured, large portions of total load remain unaccounted for. Cognitive effort, sensory intensity, environmental exposure, and autonomic stress continue to accumulate in the background, narrowing the margin between stability and collapse (Cook et al., 2017; Nijs et al., 2012; Rowe et al., 2014). At the cellular level, this threshold behavior is supported by evidence of impaired bioenergetic and signaling responses to stress. Studies in ME/CFS have demonstrated reduced efficiency of mitochondrial oxidative phosphorylation, altered ATP production dynamics, and a shift toward less efficient metabolic pathways under exertion, suggesting that cells are operating closer to their energetic limits even at baseline (Fluge et al., 2016; Tomas et al., 2017; Morris and Maes, 2014). This reduced energy availability increases vulnerability to even modest increases in demand, contributing to the disproportionate response seen in PEM.
In parallel, oxidative and nitrosative stress pathways appear to be upregulated, leading to cellular damage and further impairing mitochondrial function and signaling efficiency (Maes et al., 2011; Jammes et al., 2012). Immune activation compounds this effect. Abnormal cytokine responses following exertion, including elevations in pro-inflammatory mediators such as IL-1β, IL-6, and TNF-α, suggest that exertion triggers a prolonged inflammatory cascade rather than a transient adaptive response (Hornig et al., 2015; Komaroff and Lipkin, 2021; Montoya et al., 2017).
Neuroinflammation adds an additional layer, with evidence of microglial activation and altered central nervous system signaling in ME/CFS patients, potentially increasing the cost of cognitive and sensory processing (Nakatomi et al., 2014; Nath, 2024). These processes collectively increase the metabolic and regulatory burden placed on the system, effectively lowering the threshold at which cumulative load produces failure.
Taken together, these cellular and molecular dynamics help explain why PEM is both delayed and prolonged. The triggering event initiates a cascade of metabolic stress, immune activation, and regulatory dysfunction that unfolds over time, rather than resolving immediately after the activity ends. Recovery, therefore, requires not just rest, but re-stabilization of multiple interconnected systems, which may be impaired or slowed in affected individuals. This is the point where many patients and clinicians become misaligned with the biology of the illness. Activities that appear to be “rest” in a conventional sense may still carry significant load within one or more domains.
Conversation, decision-making, screen use, light exposure, or simply maintaining upright posture can all contribute meaningfully to cumulative burden. When these are not recognized as exertion, patients may unknowingly exceed threshold despite believing they are pacing appropriately (Jason et al., 2009; Davenport et al., 2019; NICE, 2021).
By moving beyond fatigue-based definitions and situating PEM within a systems framework that extends from cellular energetics to whole-system behavior, symptom patterns can be interpreted as structured physiological responses rather than variability or inconsistency. This shift is essential for accurate diagnosis, meaningful measurement, and effective intervention design. It also establishes PEM as a governing signal of system behavior, defining when the body is stable, when it is vulnerable, and when it is at risk of collapse. Without integrating this signal, any model of disease progression, intervention response, or remission remains structurally incomplete.
3. The Timing Problem: Why PEM Keeps Being Missed
One of the central reasons PEM has remained poorly understood is that it does not conform to the timing assumptions embedded in conventional medical reasoning. Most clinical frameworks are built around near-immediate cause-and-effect relationships, in which symptoms follow closely after a trigger. PEM frequently violates this assumption, introducing a temporal gap between exertion and symptom expression that disrupts standard diagnostic logic (Institute of Medicine, 2015; NICE, 2021). In many patients, PEM begins 12 to 72 hours after the initiating stressor, though onset and duration vary across individuals and disease states (Davenport et al., 2019; Stussman et al., 2020). This delay makes it difficult to establish causality using short observation windows or single clinical encounters. Patients may appear stable or even improved shortly after exertion, only to experience significant deterioration later. As a result, the most diagnostically relevant phase of the illness often occurs outside the timeframe in which it is evaluated (Komaroff and Bateman, 2021).
This timing mismatch is one of the clearest reasons PEM is misunderstood in clinical practice. The problem is not simply that clinicians are unaware of PEM, but that PEM behaves in a way that ordinary clinical observation is poorly designed to detect. A patient may look well during the appointment, tolerate the exam, answer questions clearly, and still crash later from the combined load of travel, posture, conversation, sensory exposure, and cognitive demand. If the crash occurs after the visit, it may never enter the clinical record. The clinician sees the moment of apparent tolerance, while the patient lives through the delayed consequence. Standard measurement approaches further compound this problem. Laboratory tests, symptom inventories, and clinical assessments are typically designed to capture static or immediately observable abnormalities, not delayed and fluctuating physiological responses. Even when abnormalities are present, they may not align temporally with symptom escalation, leading to underrecognition or misattribution of severity (Natelson et al., 2019; Russell et al., 2016). This testing-function mismatch helps explain why patients can have normal or inconclusive results while experiencing profound real-world impairment.
Fatigue-based frameworks also distort interpretation. In conventional models, fatigue is often understood as proportional to exertion and improved by rest, conditioning, or gradual activity increases. PEM does not follow that pattern. Its delayed onset, multisystem expression, and prolonged recovery indicate a failure of regulation rather than simple energy depletion. When clinicians interpret same-day tolerance as evidence of safety, they may recommend increased activity that worsens the underlying instability (Vink and Vink-Niese, 2020; NICE, 2021).
This is why PEM requires a temporal model rather than a snapshot model. The clinically meaningful question is not only whether the patient tolerated an activity in the moment, but whether the system remained stable afterward. Without that delayed follow-up window, threshold breaches are missed, worsening is misclassified, and patients are often described as inconsistent when the actual problem is that the evaluation window is too short.
Within CYNAERA’s modeling framework, this limitation is addressed through the Symptom Cascade Flare Predictor (SymCas™), which treats PEM as a time-dependent cascade rather than a point-in-time event. As described in the CYNAERA Pathophysiology of IACCs and Best Practices for ME/CFS Clinical Trials white papers, symptom expression in ME/CFS follows identifiable temporal patterns, in which early signals precede delayed system collapse. In this model, the delay between trigger and symptom escalation is not anomalous but expected, reflecting asynchronous signaling across immune, metabolic, and autonomic domains.
Post-Exertional Malaise and the Hidden Scale of ME/CFS
Earlier prevalence estimates often failed to require or properly identify PEM, leading to systematic undercounting of affected individuals (Institute of Medicine, 2015; Jason et al., 2021). Updated modeling using CYNAERA’s US-CCUC™ framework indicates that ME/CFS affects approximately 18–26 million U.S. adults, with a broader classification burden of 27.5–34.65 million when accounting for post-infectious trajectories, underdiagnosis, and structural invisibility. These estimates reflect a shift from diagnosis-based counting toward true population burden.
Clinical Implications of Timing Misalignment: A Targeted Intervention Comparison
The impact of these timing dynamics becomes clear when examining how the same patient is managed under different models of care. Consider a patient with ME/CFS who reports intermittent “good days” and attempts to increase activity during those periods. On one such day, the patient completes a 20-minute walk and moderate cognitive tasks, reporting minimal immediate symptoms.
Under a conventional model, this response is interpreted as adequate tolerance. Activity is reinforced and gradually increased based on same-day performance. However, approximately 36 hours later, the patient develops a significant worsening of symptoms, including profound fatigue, cognitive dysfunction, orthostatic instability, and pain. Because this deterioration occurs outside the immediate observation window, it may not be attributed to the preceding activity. Over time, repeated cycles of delayed worsening can lead to cumulative destabilization. In contrast, a PEM-aware model evaluates activity across a multi-day timeframe. The delayed symptom escalation is identified as a threshold breach, indicating that cumulative system load exceeded adaptive capacity. Rather than escalating activity, the intervention is adjusted to reduce baseline load, extend recovery, and prevent repeated cascade activation. This distinction reflects a fundamental shift from static to temporal modeling of disease dynamics.
Table: Conventional vs. PEM-Aware Temporal Modeling of Disease Dynamics
Dimension | Conventional Model | PEM-Aware Model (SymCas™-Aligned) |
Assessment Window | Same-day response | 24–72+ hour longitudinal tracking |
Interpretation of Initial Response | Tolerated → increase activity | Provisional → requires delayed confirmation |
Recognition of Delayed Symptoms | Often missed or misattributed | Central to evaluation |
Activity Progression | Linear escalation | Adjusted based on delayed cascade patterns |
Understanding of Worsening | Inconsistency or deconditioning | Threshold breach and system overload |
Clinical Trajectory | Progressive destabilization | Stabilization and reduced crash frequency |
Research Signal Detection | High variability, weak signal | Improved alignment with biological response |
The implications extend beyond individual care. When timing is ignored, interventions may appear safe or effective in the short term while contributing to long-term deterioration. When timing is incorporated into assessment and modeling, the same interventions can be calibrated to align with underlying physiology. This reinforces that the central issue is not patient inconsistency, but a mismatch between conventional clinical frameworks and the delayed, nonlinear dynamics that define PEM.
4. PEM as a Multi-System Cascade
Post-exertional malaise is most accurately understood as a coordinated failure across interdependent biological systems rather than dysfunction within a single pathway. While abnormalities in immune signaling, metabolic function, autonomic regulation, and neuroinflammatory processes have each been documented in ME/CFS and related conditions, these findings consistently point to a broader pattern: loss of adaptive coordination under stress (Komaroff and Lipkin, 2021; Wirth and Scheibenbogen, 2021). Within CYNAERA’s Unified Network Collapse Theory (UNCT), this pattern is conceptualized as a destabilized biological network in which regulatory domains no longer operate in synchrony when exposed to increased demand. In this framework,
PEM represents the observable output of network-level failure, emerging when cumulative system load exceeds the capacity for coordinated adaptation. In a stable physiological state, stressors such as physical exertion, cognitive demand, or environmental exposure trigger integrated responses across immune, metabolic, autonomic, and neurocognitive systems. These responses are tightly regulated through feedback mechanisms that allow the organism to maintain homeostasis and recover efficiently. In ME/CFS, baseline instability disrupts these feedback loops. When demand increases, localized dysfunction propagates across interconnected domains, resulting in delayed and prolonged system collapse.
Immune Dynamics and Adaptive Failure
At the immune level, both ME/CFS and Long COVID are associated with persistent dysregulation, including altered cytokine signaling, impaired antiviral responses, and evidence of immune exhaustion (Hornig et al., 2015; Davis et al., 2023). Of particular relevance is the identification of CD8 T-cell dysfunction, characterized by reduced production of IFNγ and TNFα and diminished responsiveness to stimulation. This pattern is consistent with functional exhaustion, in which immune cells are present but unable to mount effective responses under physiological stress .
Within a systems framework, this represents a reduction in adaptive immune capacity. Under conditions of increased demand, such as exertion, the immune system is required to regulate inflammatory signaling, coordinate repair processes, and maintain homeostasis. When immune cells are functionally compromised, signaling becomes inefficient or dysregulated. Instead of resolving stress, the system may enter a state of prolonged or delayed inflammatory activation, contributing to the temporal lag and sustained symptom amplification characteristic of PEM.
UNCT formalizes this as a network fragility state, in which impaired immune responsiveness lowers the threshold for cascade activation and increases the probability that stress exposure will result in systemic failure rather than adaptive recovery.
Metabolic Constraints and Energy Failure
Metabolic dysfunction further constrains the system’s ability to respond to stress. Evidence in ME/CFS demonstrates impaired oxidative phosphorylation, reduced ATP availability, and early reliance on anaerobic metabolism, leading to increased lactate accumulation even at low levels of exertion (Naviaux et al., 2016; Tomas et al., 2017; Fluge et al., 2016). These abnormalities create a state in which energy supply cannot meet demand during stress exposure. As a result, processes that depend on adequate energy availability, including immune signaling, neural processing, and autonomic regulation, become less effective. The accumulation of metabolic byproducts and oxidative stress further disrupts cellular signaling and amplifies dysfunction across systems. Within UNCT, metabolic impairment represents a capacity constraint layer, limiting the system’s ability to sustain coordinated responses and increasing vulnerability to collapse under load.
Autonomic and Vascular Dysregulation
Autonomic dysfunction represents a third critical domain in the PEM cascade. Patients with ME/CFS frequently exhibit orthostatic intolerance, reduced heart rate variability, and abnormal sympathetic activation, all of which impair cardiovascular regulation and blood flow distribution (Rowe et al., 2014; Raj et al., 2020; Systrom et al., 2021). During exertion, these impairments limit the system’s ability to deliver oxygen and nutrients efficiently to tissues, particularly the brain and skeletal muscle. Reduced cerebral perfusion contributes to cognitive dysfunction, while impaired peripheral circulation exacerbates physical fatigue and metabolic stress. Autonomic dysregulation also affects neuroimmune communication, further linking this domain to immune and metabolic dysfunction. In UNCT terms, autonomic instability functions as a coordination disruption layer, impairing the system’s ability to synchronize responses across domains.
Neuroinflammatory and Central Processing Effects
Neuroinflammatory processes add an additional layer of complexity to the PEM cascade. Imaging studies have demonstrated evidence of microglial activation and central nervous system inflammation in ME/CFS, which may underlie cognitive dysfunction, sensory hypersensitivity, and impaired stress tolerance (Nakatomi et al., 2014; Nath, 2024). These processes affect how the central nervous system processes and responds to physiological stress, potentially lowering tolerance thresholds and amplifying symptom responses. They also interact with autonomic and immune pathways, reinforcing the interconnected nature of the system. Within UNCT, neuroinflammation contributes to signal amplification and threshold sensitivity, increasing the likelihood that stress exposure will trigger disproportionate system-wide responses.
Integrated Cascade Model
These domains do not operate independently. Instead, they form a dynamic network in which dysfunction in one domain amplifies instability across others. Immune dysregulation can drive metabolic inefficiency; metabolic stress can amplify inflammatory signaling; autonomic instability can impair both vascular and immune coordination; and neuroinflammation can disrupt system-wide regulation. Under this model, PEM occurs when a stressor increases demand across these interconnected systems beyond their capacity for coordinated adaptation. The resulting cascade unfolds over time, consistent with the delayed onset of symptoms, and persists due to impaired recovery mechanisms. This relationship can be summarized as follows:
Table: Multi-System Cascade Dynamics in PEM
Domain | Observed Dysfunction | Network-Level Role (UNCT) | Contribution to PEM |
Immune | CD8 T-cell dysfunction, altered cytokines | Reduced adaptive capacity (network fragility) | Delayed inflammatory signaling, poor stress resolution |
Metabolic | Impaired ATP production, lactate accumulation | Capacity constraint layer | Energy shortfall under load, metabolic stress amplification |
Autonomic / Vascular | Orthostatic intolerance, low HRV | Coordination disruption layer | Impaired oxygen delivery, instability during exertion |
Neuroinflammatory | Microglial activation, cognitive dysfunction | Signal amplification layer | Lowered tolerance threshold, sensory and cognitive PEM |
Redox / Oxidative Stress | Elevated ROS/RNS | Cross-domain destabilization | Reinforces immune and metabolic dysfunction loops |
Within this framework, PEM is not the result of a single failing system, but the emergent property of a destabilized biological network. Its severity, timing, and duration reflect the degree of underlying system instability and the extent to which coordination across domains has been compromised.
5. The Many Forms of Exertion: Redefining Load in PEM
A central limitation in traditional interpretations of post-exertional malaise is the narrow definition of exertion as physical activity alone. While physical exertion is a well-recognized trigger, clinical observation and emerging research demonstrate that PEM can be initiated by a wide range of stressors that extend beyond muscular effort (Carruthers et al., 2011; Jason et al., 2015; Davenport et al., 2019). Within CYNAERA’s modeling framework, exertion is more accurately conceptualized as total system load, defined as the cumulative demand placed on immune, metabolic, autonomic, neurocognitive, and sensory systems over time. PEM occurs when this cumulative load exceeds the system’s reduced capacity for coordinated adaptation, consistent with threshold-based failure dynamics described in earlier sections. This broader definition aligns with findings from CYNAERA’s Game Terrain Diagnostics™ framework, which demonstrates that functional decline emerges under cognitive, sensory, and decision-making load in real-world environments, often before overt physical limitation is apparent . These findings reinforce that PEM is not tied to a single domain of exertion, but to integrated system demand across multiple domains simultaneously.
Cognitive Load as a Primary Driver of PEM
Cognitive exertion is a common and frequently underrecognized trigger of PEM. Tasks involving sustained attention, executive function, memory processing, decision-making, or multitasking can produce significant physiological strain, even in the absence of physical activity. Neuroimaging and clinical studies suggest that ME/CFS is associated with neuroinflammation, impaired cerebral perfusion, and altered neural efficiency, all of which increase the energetic cost of cognitive processing (Nakatomi et al., 2014; Nath, 2024; Systrom et al., 2021). As a result, activities that would be considered low-demand in healthy individuals may exceed cognitive capacity in affected patients. Game Terrain Diagnostics™ extends this understanding by demonstrating that cognitive load manifests as measurable performance drift, including increased reaction time variability, reduced multitasking tolerance, and early task abandonment . These behavioral adaptations reflect underlying physiologic strain and often precede overt symptom escalation In this context, cognitive exertion should be treated as equivalent to physical exertion in its ability to trigger PEM.
Sensory Load and Neurophysiologic Amplification
Sensory input represents another major contributor to system load. Patients with ME/CFS frequently report hypersensitivity to light, sound, temperature, and chemical exposures, suggesting altered central processing and increased neurophysiologic cost of sensory integration (Nijs et al., 2012; Afrin et al., 2020). High sensory environments, such as crowded spaces, bright screens, or complex visual stimuli, can increase neural demand and contribute to cumulative system load. Within Game Terrain Diagnostics™, sensory load is reflected in changes in interaction patterns under high-input conditions, including reduced session duration and increased variability in performance . These findings suggest that sensory exposure is not a passive experience, but an active contributor to physiological strain, capable of triggering or amplifying PEM.
Orthostatic and Autonomic Load
Orthostatic stress represents a third major category of exertion. Activities such as standing, sitting upright, or transitioning between postures require continuous autonomic regulation to maintain blood pressure, heart rate, and cerebral perfusion. In patients with dysautonomia, including POTS, these processes are impaired, leading to increased physiological cost during upright activity (Rowe et al., 2014; Raj et al., 2020). Even low-intensity tasks performed in an upright position may significantly increase system load. This form of exertion is often overlooked because it is not perceived as “activity” in the traditional sense. However, from a systems perspective, orthostatic demand contributes directly to cumulative load and may interact with cognitive and sensory stressors to trigger PEM.
Emotional and Neuroendocrine Load
Emotional stress, while often mischaracterized in purely psychological terms, has measurable physiological effects mediated through neuroendocrine and autonomic pathways. Activation of stress-response systems increases heart rate, alters cortisol dynamics, and influences immune signaling, all of which contribute to system load (Natelson et al., 2019). In individuals with underlying instability, these responses may be amplified or poorly regulated, increasing vulnerability to threshold breaches. Emotional stress therefore functions as a physiological input that can contribute to PEM, particularly when combined with other forms of load.
Environmental Load and Real-World Conditions
Environmental conditions such as heat, humidity, air quality, and chemical exposure further modulate system load. These factors can influence autonomic regulation, inflammatory signaling, and metabolic demand, particularly in patients with heightened sensitivity (NICE, 2021; Komaroff, 2019). As demonstrated in CYNAERA’s real-world modeling frameworks, environmental variability can significantly alter functional capacity on a day-to-day basis. This reinforces that PEM cannot be fully understood or managed without accounting for environmental context.
Integrated Load Model
Taken together, these findings support a unified model in which PEM is driven by cumulative, multi-domain load rather than isolated exertion. Each domain contributes to total system demand, and interactions between domains amplify overall stress.
Table: Multi-Domain Load Contributions to PEM
Load Domain | Examples | Underlying Mechanism | Observed Signal (CYNAERA) | Contribution to PEM |
Physical | Walking, exercise | Energy demand, metabolic strain | Reduced endurance, delayed crash | Direct threshold contribution |
Cognitive | Problem-solving, multitasking | Neuroinflammation, perfusion deficits | Reaction time variability, task simplification | Early instability + delayed crash |
Sensory | Light, sound, screens | Central sensitization | Session shortening, variability spikes | Amplifies total load |
Orthostatic | Standing, sitting upright | Autonomic dysfunction | Reduced tolerance, fatigue onset | Continuous background load |
Emotional | Stress, conflict | Neuroendocrine activation | Variability under pressure | Adds to cumulative load |
Environmental | Heat, pollution, mold | Inflammatory + autonomic stress | Day-to-day capacity shifts | Modulates threshold dynamically |
Within this framework, PEM is not triggered by a single form of exertion, but by the accumulation of demand across multiple domains. CYNAERA modeling demonstrates that functional decline often emerges first under combined cognitive, sensory, and environmental load, reinforcing that PEM is a systems-level response to integrated stress rather than isolated activity.
6. Why Legacy Research and Clinical Models Keep Getting PEM Wrong
Despite decades of patient reporting and formal recognition of post-exertional malaise, PEM remains poorly integrated into both clinical practice and research design. This gap does not reflect a lack of evidence, but a persistent mismatch between the properties of PEM and the frameworks used to study it. At its core, PEM is a delayed, multi-system, threshold-dependent phenomenon. Legacy medical models, by contrast, are built around assumptions of immediacy, linearity, and relative uniformity of response. Those assumptions work in conditions where symptom change occurs close to the triggering event and where intervention effects can be captured through fixed measurement windows. They fail in ME/CFS because PEM does not behave like a linear symptom. It behaves like a cascade.
Traditional clinical care still relies heavily on static observations. Patients are assessed in clinic, asked how they feel in that moment, and evaluated using tools designed to capture symptoms or abnormalities within narrow timeframes. Yet PEM frequently unfolds outside those windows. A patient may appear stable or even improved during the visit and then deteriorate significantly 24 to 72 hours later. The more the system depends on snapshot measurement, the more likely it is to miss the defining feature of the illness (Institute of Medicine, 2015; Komaroff and Bateman, 2021; Stussman et al., 2020).
This misalignment becomes especially consequential when clinicians interpret immediate tolerance as meaningful tolerance. If a patient completes an activity, tolerates an appointment, or initiates an intervention without same-day worsening, the response may be interpreted as evidence of safety or progress. In PEM-driven disease, that interpretation is often incorrect. Same-day tolerance may simply reflect a system that has not yet declared its failure state. CYNAERA’s SymCas™ framework addresses this limitation by modeling PEM as a longitudinal cascade in which early instability signals precede delayed collapse. Within this model, variability is not noise but an early indicator of threshold breach. Without incorporating temporal dynamics, clinical interpretation remains systematically misaligned with underlying physiology.
The Hidden High-Risk Group: Super-Sensitive Patients
A major failure of legacy models is their inability to recognize and safely manage super-sensitive patients, a subgroup that is especially common across ME/CFS, Long COVID, POTS, MCAS, and related conditions. These patients are not simply more symptomatic. They are biologically less buffered. Their systems operate with reduced tolerance for physiologic, pharmacologic, sensory, and environmental input. As a result, exposures that appear trivial in other populations may trigger disproportionate destabilization. This group frequently demonstrates:
reactivity to low-dose medications or supplements
sensitivity to excipients, chemicals, temperature, or air quality
amplified responses to cognitive, orthostatic, or emotional load
prolonged and more severe PEM following relatively minor triggers
In conventional care, these patterns are often misinterpreted as anxiety, somatic amplification, or noncompliance. In reality, they reflect measurable system fragility. CYNAERA’s MCAS-sensitive screening logic helps identify this subgroup using structured indicators such as medication sensitivity, environmental reactivity, autonomic overlap, and post-infectious onset patterns . These are not incidental findings. They are signals that the patient’s threshold for destabilization is significantly reduced.
Why STAIR™ Is Required for Interpretability
This is the context in which STAIR™ (Stabilization, Tolerance, and Immune Readiness) becomes essential. Conventional models assume that intervention can begin immediately and that patient response will reveal efficacy. For super-sensitive and unstable patients, this assumption breaks down. The response observed may reflect the system’s instability rather than the intervention itself.
STAIR™ reframes the problem by introducing readiness as a prerequisite for interpretation. The key question shifts from “Does the patient tolerate the intervention?” to “Was the system stable enough for that tolerance to be meaningfully assessed?” In practice, this includes:
identifying hypersensitivity and mast cell instability
reducing environmental and inflammatory load prior to intervention
avoiding exposure during active flare states
using staged or micro-dose escalation rather than standard dosing
These adjustments are not precautionary. They are required to prevent the introduction of new load into a system already operating near threshold. Without this step, destabilization is likely and frequently misinterpreted as treatment failure or intolerance. Super-sensitive patients make this failure mode visible. They do not create the problem. They reveal it.
The Missing Layer: Phenotyping and Diagnostic Fingerprinting
A second structural failure is the persistent treatment of ME/CFS as a relatively uniform condition. In reality, patients differ significantly across immune, autonomic, metabolic, endocrine, and environmental domains. CYNAERA’s phenotyping framework organizes these differences into structured subtypes, allowing PEM expression to be interpreted in relation to underlying system configuration. A patient with autonomic-dominant dysfunction may experience orthostatic-triggered PEM, while another with immune reactivation may exhibit delayed inflammatory crashes. Without stratification, these patterns are collapsed into a single category, obscuring mechanism and response.
The Composite Diagnostic Fingerprint (CDF-ME/CFS™) extends this logic by assigning a multi-domain profile that integrates symptom patterns, trigger sensitivity, physiologic instability, and comorbid conditions. This allows threshold behavior, intervention tolerance, and recovery dynamics to be interpreted in a structured way. Without phenotyping and CDF:
heterogeneous patients are grouped together, masking true response patterns
sensitive subgroups are destabilized or excluded
variability is misinterpreted as inconsistency
trial outcomes reflect noise rather than signal
When phenotype and fingerprinting are applied, variability becomes interpretable. Differences in response can be traced to underlying system structure rather than dismissed as randomness.
The Final Gap: The Absence of a Coherent Remission Standard
Even when timing, sensitivity, and phenotype are partially accounted for, a final gap remains: the absence of a consistent and biologically accurate definition of remission. Across ME/CFS and related conditions, remission is often inferred from symptom reduction or short-term improvement. These definitions assume that visible improvement reflects underlying recovery. In PEM-driven disease, that assumption is frequently incorrect. Patients may demonstrate functional gains while remaining highly vulnerable to relapse under exertion or environmental stress (Komaroff and Bateman, 2021; Nasserie et al., 2021). This creates a systematic distortion. Improvement without stability is treated as success, while delayed relapse is treated as unpredictability.
The CYNAERA Remission Standard™ addresses this by defining remission as a state-dependent, multi-domain outcome based on stability, durability, function, flare control, and resilience . Within this model, remission is not a moment. It is a pattern that must hold under real-world conditions.
This is particularly important in PEM. A patient who reports reduced fatigue but continues to crash after exertion has not achieved remission in a systems sense. They have entered a partially stabilized state with persistent vulnerability.
Without a structured remission framework:
short-term improvement is overinterpreted
relapse dynamics are underweighted
treatment durability is misclassified
trial outcomes become inconsistent
With it, improvement can be graded, compared, and understood in relation to system behavior.
Why Trials Keep Repeating the Same Mistake
These failures become most visible in clinical trials, where the architecture itself assumes the wrong disease model. Standard trial designs presume:
stable baseline conditions
uniform tolerance across participants
linear dose-response relationships
These assumptions do not hold in PEM-driven illness. As CYNAERA’s trial analysis demonstrates, many ME/CFS trials have failed not because therapies were ineffective, but because trial design was misaligned with patient biology . Delayed PEM responses fall outside endpoint windows. Sensitive patients destabilize under standard protocols. Heterogeneous subgroups dilute signal. The recurring structural failures include:
static measurement in a disease defined by delayed and fluctuating response
same-day interpretation of tolerance despite delayed collapse dynamics
inadequate subgrouping across immune, autonomic, and sensitivity phenotypes
failure to distinguish unstable terrain from true nonresponse
exposure of highly sensitive patients without stability gating
From Failure to Alignment: A Systems-Based Correction
The failures observed in ME/CFS care and research are not random. They are the predictable result of applying static, uniform models to a dynamic, heterogeneous condition.
A systems-based correction requires alignment across four dimensions:
temporal modeling of delayed symptom dynamics (SymCas™)
stability-first intervention gating (STAIR™)
phenotype and fingerprint-based stratification (CDF-ME/CFS™)
state-dependent outcome measurement (Remission Standard™)
When these elements are integrated, PEM becomes interpretable rather than inconsistent, and treatment response becomes measurable rather than ambiguous. The persistent failure to understand PEM is not due to its complexity alone, but to continued reliance on frameworks that assume a simpler illness than the one patients actually have. Without accounting for delayed kinetics, system sensitivity, phenotype variation, and state-dependent remission, clinical and research systems will continue to misclassify both improvement and failure. Until these dimensions are integrated, the patients most affected will remain both the most harmed and the least accurately measured.
7. PEM as a Threshold Failure Model
Post-exertional malaise can be most accurately understood as a threshold-dependent failure of system-wide adaptive capacity, in which cumulative physiological demand exceeds the body’s ability to maintain coordinated function across immune, metabolic, autonomic, and neurocognitive domains. This interpretation aligns with decades of work describing ME/CFS as a multi-system illness rather than a disorder of fatigue alone, including foundational definitions from the Institute of Medicine (2015) and the International Consensus Criteria led by Carruthers and colleagues (Carruthers et al., 2011).
Clinicians and researchers such as Anthony Komaroff and W. Ian Lipkin have emphasized that ME/CFS reflects complex neuroimmune and metabolic dysfunction, rather than simple deconditioning (Komaroff and Lipkin, 2021). Similarly, Carmen Scheibenbogen and Klaus Wirth have proposed mechanistic models involving vascular dysregulation and impaired oxygen delivery, further supporting the idea that exertional intolerance reflects systemic constraint rather than isolated fatigue (Wirth and Scheibenbogen, 2021). Within this context, the threshold model resolves a long-standing clinical paradox. Patients consistently report that the same activity may be tolerated on one day and trigger severe worsening on another, and that minimal cognitive, sensory, or orthostatic demand can provoke disproportionate collapse. Work by Staci Stevens and Christopher Snell using cardiopulmonary exercise testing (CPET) has shown objective reductions in functional capacity on repeat exertion, supporting the idea that PEM reflects a failure of recovery rather than simple exertion intolerance (Snell et al., 2013; VanNess et al., 2010). These observations are difficult to reconcile within linear models, but are expected in a system where state determines response.
Threshold Capacity as a Function of System State
Within this framework, the PEM threshold is dynamic. It shifts according to the patient’s biological state, cumulative exposure burden, recovery status, and underlying phenotype.
This relationship can be expressed conceptually as:
Threshold Capacity = f (Stability + Phenotype + Recovery State − Total Load)
Each component is supported by existing research:
Stability reflects current regulation across immune, autonomic, and metabolic systems. Work by Nancy Klimas, Maureen Hanson, and Mady Hornig has demonstrated persistent immune dysregulation and altered cytokine signaling in ME/CFS, indicating that baseline state is often already destabilized (Hornig et al., 2015; Klimas et al., 2012; Hanson, 2016).
Phenotype reflects structured differences in disease expression. Leonard Jason’s work on case definitions and subgrouping highlights the heterogeneity within ME/CFS populations, reinforcing the need for stratification rather than uniform classification (Jason et al., 2015).
Recovery State reflects delayed kinetics of symptom expression. Research by Todd Davenport
and colleagues has shown that PEM onset and duration vary significantly and are often delayed, confirming that recovery cannot be inferred from immediate response (Davenport et al., 2019).
Total Load reflects cumulative multi-domain demand. Studies on cognitive exertion (Cook et al., 2017), orthostatic intolerance (Rowe et al., 2014), and sensory hypersensitivity (Nijs et al., 2012) demonstrate that exertion in ME/CFS extends beyond physical activity alone.
This formulation is intentionally presented as a conceptual systems model rather than a finalized clinical equation. Its purpose is to organize converging evidence into a framework that explains why PEM is delayed, nonlinear, and state-dependent.
Why Threshold Variability Appears Unpredictable
A defining feature of PEM is its apparent unpredictability. Patients frequently report that tolerance fluctuates day to day, or that minor activities trigger major crashes. Within a threshold model, this variability reflects unmeasured shifts in system state rather than randomness. Research has identified multiple contributors to these shifts, including immune activation (Hornig et al., 2015), metabolic dysfunction (Naviaux et al., 2016; Fluge et al., 2016), impaired oxygen extraction (Systrom et al., 2021), and autonomic instability (Raj et al., 2020).
Ron Davis and colleagues at Stanford have also highlighted metabolic and cellular stress abnormalities in ME/CFS, suggesting that even minor perturbations may overwhelm already compromised systems. When combined with environmental and behavioral factors, these shifts continuously alter threshold capacity. Because clinical and research systems rarely measure these variables simultaneously, threshold movement appears inconsistent. In reality, it reflects untracked changes in biologic conditions interacting over time.
PEM as a Conversion Event Rather Than a Simple Crash
The threshold model aligns with CYNAERA’s Primary Chronic Trigger (PCT) framework, in which PEM functions as a conversion event linking acute stress exposure to longer-term system destabilization. This interpretation is consistent with literature describing post-infectious illness progression. Studies of ME/CFS and Long COVID by Akiko Iwasaki, David Putrino, and others have shown that immune dysregulation, viral persistence, and autonomic dysfunction may persist long after initial infection, with symptom flares reflecting underlying system instability (Davis et al., 2023; Peluso et al., 2024).
Within this context:
a trigger increases system demand
a threshold breach represents failure to recover
repeated breaches may progressively reduce baseline capacity
This helps explain why early stabilization is associated with better outcomes, and why repeated PEM episodes are often linked to long-term functional decline (Institute of Medicine, 2015; Tomas et al., 2017).
Phenotype-Driven Threshold Behavior
Threshold behavior varies significantly across patients. Research on dysautonomia led by Peter Rowe and Satish Raj demonstrates that autonomic dysfunction alone can produce substantial exertional intolerance (Rowe et al., 2014; Raj et al., 2020). Meanwhile, mast cell–related work by Lawrence Afrin and colleagues highlights how immune hypersensitivity can drive exaggerated responses to environmental and pharmacologic triggers (Afrin et al., 2020). Metabolic research by Fluge and Mella, as well as Naviaux, supports the existence of energy production abnormalities that may limit tolerance even at low levels of exertion (Fluge et al., 2016; Naviaux et al., 2016). These differences reinforce that threshold behavior is phenotype-dependent. Without structured stratification, these patterns are collapsed, making PEM appear inconsistent.
Super-Sensitive Patients and Threshold Compression
Super-sensitive patients represent an extreme form of threshold compression. These individuals often exhibit overlapping features described in MCAS, dysautonomia, and ME/CFS literature, including heightened reactivity to medications, environmental triggers, and physiologic stress (Afrin et al., 2020; Raj et al., 2020). Their systems operate with minimal buffering capacity, meaning that even low-level inputs may trigger disproportionate responses. Within a STAIR™ framework, these patients are best understood as operating near continuous threshold proximity. Their responses are not exaggerated but reflect high-resolution manifestations of system instability.
Threshold Expansion as the Basis of Remission
The threshold model provides a clearer definition of recovery. Rather than focusing on symptom reduction alone, remission must reflect increased system capacity and improved tolerance to load. This aligns with CYNAERA’s Remission Standard™, which evaluates stability, durability, function, flare control, and resilience. This perspective is consistent with broader chronic illness research emphasizing sustained control rather than isolated improvement, including treat-to-target strategies in autoimmune disease (Smolen et al., 2023). A patient who remains vulnerable to collapse under modest load has not achieved remission, regardless of short-term symptom improvement.
From Observation to Prediction
One of the most important implications of this model is that PEM can move from description to prediction. When threshold capacity is understood as a function of system state, and when load is tracked across domains, it becomes possible to:
identify conditions under which threshold breach is likely
detect early instability signals
define safer windows for intervention
interpret variability as structured rather than random
This aligns with emerging trends in precision medicine and systems biology, which emphasize dynamic modeling over static classification. PEM is not an exaggerated response to exertion. It is a system-level failure that occurs when cumulative demand exceeds a dynamically shifting threshold of tolerance. When grounded in existing immunologic, metabolic, autonomic, and neuroinflammatory research, and structured through a systems-based framework, PEM becomes not only biologically coherent, but measurable, interpretable, and increasingly predictable.
8. From Threshold Failure to Stabilization: A Systems-Based Intervention Model
If post-exertional malaise (PEM) reflects a threshold-dependent failure of system-wide adaptive capacity, then any intervention model that focuses exclusively on physical exertion is structurally incomplete. This is not a matter of opinion or preference in management style. It is a direct consequence of how the system behaves. PEM is not triggered by a single input variable. It emerges from the interaction between cumulative load, threshold sensitivity, and recovery dynamics across multiple physiological and cognitive domains.
Current clinical guidance often centers on reducing activity, avoiding overexertion, and, in more advanced cases, pacing based on heart rate or perceived exertion. While these strategies represent meaningful progress compared to earlier recommendations that encouraged graded increases in activity, they still operate within a narrowed definition of exertion. The implicit assumption is that exertion is primarily physical, and that by controlling physical output, the system can be stabilized. For many patients, this assumption proves insufficient. They reduce walking, eliminate exercise, limit household tasks, and still experience instability that appears disproportionate to what they have done.
This is the point at which patients begin to feel as though their illness is inconsistent, unpredictable, or resistant to intervention. In reality, the system is behaving consistently. The mismatch lies in what is being measured. What has been missing is not effort, discipline, or adherence to medical advice. What has been missing is a complete accounting of load. Patients are rarely taught to recognize that cognitive effort, sensory processing, posture-related autonomic demand, environmental stressors, and emotional engagement all draw from the same limited adaptive reserve. When these domains are not identified as exertion, they continue to accumulate in the background, even as visible physical activity is reduced. The system responds to the total, not the subset that is easiest to observe. This is why a patient can experience what appears to be a contradiction. They do less, yet do not improve. They rest, yet still crash. They follow guidance, yet remain unstable. The contradiction is not in the patient’s experience. It is in the model being used to interpret that experience.
Heart Rate Thresholding as a Foundational but Incomplete Model
The introduction of heart rate–based pacing marked a critical advancement in the management of ME/CFS and related conditions. Drawing on CPET research by Christopher Snell, Staci Stevens, and Mark VanNess, this approach operationalized the concept of an anaerobic threshold and made it accessible for daily use. By applying formulas such as:
Estimated Anaerobic Threshold = (220 − age) × 0.6 Severe / Conservative Threshold = (220 − age) × 0.5
patients gained a practical tool for limiting cardiometabolic exertion and reducing the likelihood of triggering PEM (Snell et al.; Stevens et al.). The significance of this shift cannot be overstated. It established that PEM behaves in a threshold-dependent manner and that crossing that threshold carries consequences that are often delayed, nonlinear, and prolonged. It moved the field away from vague advice and toward something that could be measured, tracked, and adjusted.
However, the strength of heart rate pacing also introduced a subtle limitation. Because it effectively captures one major dimension of exertion, it is often interpreted as representing total system load. In reality, it reflects a specific subset of physiological demand, primarily cardiovascular and metabolic strain. It does not directly capture the cost of sustained cognitive processing, the impact of sensory variability, the autonomic burden of posture, or the influence of environmental and emotional factors. This distinction is not theoretical. It is observable in patient outcomes. Patients frequently report that they remain within their heart rate limits, adhere strictly to pacing guidelines, and still experience crashes that appear disconnected from their activity. From the perspective of a single-domain model, this can seem inconsistent or even inexplicable. From a multi-domain model, it is expected.
The implications of this chart are not trivial. They reveal why heart rate pacing, while effective, cannot fully stabilize the system on its own. It reduces one major source of threshold pressure while leaving several others unmeasured. When those unmeasured domains remain active, the system can still accumulate sufficient load to trigger PEM. This is the point where many patients become trapped in a cycle of partial improvement and recurrent destabilization. They are using a valid tool, but applying it within an incomplete model. The issue is not misuse. It is scope.
The Total Load Problem
To resolve this limitation, load must be conceptualized as a cumulative, multi-domain variable rather than a single observable input. This can be expressed as:
Load_total = Physical + Cognitive + Sensory + Autonomic + Environmental + Emotional
or in compact form:
Load_total = Lp + Lc + Ls + La + Le + Lm
Each component represents a distinct domain of demand, and importantly, these domains do not operate independently. They interact. Cognitive effort can increase autonomic activation. Sensory overload can amplify emotional stress. Postural demands can alter cerebral perfusion. Environmental conditions can reduce the system’s overall tolerance. The combined effect is not simply additive in all cases. It can be multiplicative, particularly in an already sensitized system.
Existing research supports the role of cognitive exertion, sensory input, and orthostatic stress in provoking PEM (Cook et al., 2017; Davenport et al., 2019; Nijs et al., 2012; Rowe et al., 2014). The contribution of this framework is not to introduce entirely new domains, but to organize them into a structure that can be used for intervention.
This reframing clarifies a key clinical observation. Patients are not necessarily exceeding threshold because they performed a single high-intensity task. More often, they exceed threshold because they accumulated moderate demand across multiple domains that were never classified as exertion. This is the structural explanation behind statements such as “I didn’t do much, but I still crashed.” The system is responding to total load, not visible activity.
Comparative Framing: Why Heart Rate Pacing Alone Plateaus
The limitations of single-domain pacing become clearer when pacing strategies are compared directly.
Table. Comparative Model of Pacing Architectures in ME/CFS
Dimension | Heart Rate–Only Pacing | Multi-Domain Pacing (HR + Cognitive + Environmental) |
Primary Target | Physical exertion | Total system load |
Mechanism | Maintains activity below anaerobic threshold | Matches demand to capacity across domains |
Captures Cognitive Load | No | Yes |
Captures Sensory Load | No | Yes |
Hidden Load | High | Reduced |
Threshold Breach Risk | Partial | Broad reduction |
Flare Frequency | Moderate | Lower |
Recovery Efficiency | Variable | Improved |
Stability Trajectory | Plateau or fluctuation common | More consistent stabilization |
False Stability Risk | High | Lower |
This comparison illustrates a central point. Heart rate pacing reduces the likelihood of threshold breach from physical exertion, but it does not eliminate threshold pressure from other domains. As a result, patients may experience fewer immediate crashes related to overt activity while continuing to accumulate load in less visible ways. This creates a pattern that can be described as “false stability,” where the system appears controlled in the short term but remains fundamentally unstable. From a clinical perspective, this distinction is critical. When patients plateau under heart rate pacing, it is often interpreted as a limitation of the intervention or as evidence of disease severity. A more accurate interpretation is that the intervention is incomplete. Once additional domains are incorporated, the persistence of symptoms becomes more understandable, and further stabilization becomes possible.
A Dynamic Stability Model
To fully capture the behavior of the system, stability must be modeled as a function that evolves over time rather than a static condition.
Stability(t+1) = Stability(t) + R(t) − [Load_total × S(t)] − F(t+Δ)
Where:
R(t) represents recovery gain
S(t) represents threshold sensitivity
F(t+Δ) represents delayed flare penalty
The delayed component is essential. In many patients, the consequences of threshold breach do not manifest immediately. Instead, they emerge hours or days later, often obscuring the relationship between cause and effect. The flare term can be expanded conceptually as:
F(t+Δ) = ∫ (Load_total × S) dt over prior window
This formulation captures a lived reality for patients. The crash is not always caused by the most recent activity. It is often the result of accumulated load interacting with a system whose tolerance has already been reduced. This is why patients can experience severe deterioration after what appears to be minimal activity. The system is responding to cumulative burden, not isolated events.
Comparative Intervention Modeling: Different Pacing Architectures
The practical implications of this model can be illustrated by comparing system behavior under different pacing strategies. Under heart rate–only pacing:
Load_total ≈ Lp (managed) + Lc + Ls + La + Le + Lm
Physical exertion is controlled, but background load remains active. The resulting system behavior is characterized by oscillation, delayed destabilization, and incomplete recovery.
Under multi-domain pacing:
Load_total ≈ Lp↓ + Lc↓ + Ls↓ + La↓ + Le↓ + Lm↓
Both peak and baseline load are reduced. This does not eliminate variability, but it alters its structure. Fluctuations become narrower, delayed crashes become less frequent, and recovery gains are more likely to accumulate. The distinction is structural rather than incremental. Heart rate pacing reduces one major driver of instability. Multi-domain pacing reduces the cumulative system burden that produces instability. This is the transition point in the model. Once load is understood in this way, the focus of intervention shifts from limiting visible exertion to aligning total demand with available capacity across domains. That shift is what allows the discussion to move from pacing as damage control to pacing as a pathway toward stabilization and, eventually, remission.
Operationalizing Multi-Domain Load: From Conceptual Model to Clinical Use
Once load is defined as a multi-domain construct and stability is modeled as a time-dependent function, the remaining challenge is translation. The field does not suffer from a lack of awareness that patients experience cognitive fatigue, sensory intolerance, and autonomic instability. What it lacks is a coherent method for incorporating those domains into daily decision-making in a way that is both clinically interpretable and practically usable. Without that translation layer, pacing remains conceptually correct but operationally incomplete.
The Branching Load Index (BLI) and Game Cognitive Load Rating (GCLR) frameworks address this gap by separating the system into two interacting variables: available capacity and task demand. This distinction aligns with broader models of physiologic stress and adaptation, including allostatic load theory and energy envelope models, which describe how cumulative burden across domains leads to system destabilization when adaptive reserves are exceeded (McEwen, 1998; Jason et al., 2009). In this formulation, BLI represents the system’s available adaptive reserve at a given point in time, shaped by prior exertion, sleep quality, autonomic tone, inflammatory state, and environmental exposures. GCLR represents the structural demand imposed by a task, decomposed into components that reflect how the nervous system processes and responds to that task under real-world conditions. This relationship can be expressed as:
GCLR_eff = Σ(factor scores × weights) − Σ(adjustments) Risk = GCLR_eff − BLI + Environmental Penalty
Extending this into a probabilistic framework:
p(flare) = 1 / (1 + e^-(α + β × Risk))
While the mathematical representation is conceptual rather than prescriptive, it reflects a well-established principle across physiology and behavioral science: outcomes are determined not by absolute demand or absolute capacity, but by the gap between them. Similar formulations appear in fatigue modeling, cognitive load theory, and occupational stress research, where performance degradation and failure risk increase as demand exceeds available resources (Hockey, 2013; Boksem & Tops, 2008). In the context of ME/CFS and related conditions, this gap manifests as post-exertional symptom exacerbation, delayed recovery, and progressive instability.
The importance of this framework lies in its ability to explain variability that is otherwise interpreted as inconsistency. A task that is tolerated on one day may trigger a flare on another, not because the task itself changed, but because the system’s capacity entering the task was different. This aligns with findings in ME/CFS and Long COVID showing fluctuating autonomic function, impaired oxygen extraction, and variable cerebral perfusion, all of which alter the system’s tolerance for both physical and cognitive load (Systrom et al., 2022; van Campen et al., 2020; Dani et al., 2021). By explicitly modeling this interaction, the framework provides a mechanism for understanding day-to-day variability without attributing it to randomness.
Cognitive Load as a Primary Driver of Instability
Among the domains contributing to total load, cognitive demand remains the most consistently underestimated. This underestimation is partly cultural and partly structural. Cognitive effort is often not classified as exertion because it does not produce the same visible markers as physical activity. However, research across neurophysiology and cognitive science demonstrates that sustained cognitive processing carries measurable metabolic cost, increases neural energy demand, and interacts with autonomic regulation (Raichle & Mintun, 2006; Cook et al., 2017). In ME/CFS and Long COVID, these effects are amplified by underlying abnormalities in energy metabolism, neuroinflammation, and cerebral blood flow (Komaroff & Lipkin, 2021; Nakatomi et al., 2014; Systrom et al., 2022).
Cognitive load is not a single variable but a composite of interacting demands. These include processing speed requirements, task complexity, working memory load, attentional switching, sensory integration, and the degree of precision required for task completion. When these factors are combined, the resulting demand can exceed what is suggested by the outward appearance of the activity. For example, tasks involving high switching frequency and sustained working memory demands have been shown to produce disproportionate fatigue and performance decline, even in otherwise healthy populations (Monsell, 2003; Unsworth & Robison, 2017). In individuals with impaired energy metabolism or autonomic dysfunction, the threshold for these effects is significantly lower. This helps explain why patients can tolerate certain forms of low-complexity activity while experiencing severe consequences from tasks that are cognitively dense but physically minimal. It also clarifies why improvements in physical tolerance do not necessarily translate into improvements in overall stability. If cognitive load remains high, the system continues to operate near threshold, preventing recovery from consolidating.
Reframing “Rest” Through Load Architecture
This is the point in the model where misunderstanding has the greatest clinical impact, and where correcting that misunderstanding produces the most immediate change in patient outcomes. In both clinical guidance and everyday language, rest is typically defined in behavioral terms, as the absence of visible exertion. Sitting, watching something, reading, or engaging in a calm activity is generally interpreted as rest. Within a multi-domain load system, that definition is insufficient.
Rest must instead be defined physiologically, as a state in which total system demand is reduced to a level that allows recovery processes to exceed ongoing burden. This distinction is not semantic. It explains one of the most consistent and distressing patterns reported by patients: the experience of “resting” followed by delayed collapse.
Chart: Reframing “Rest” Through Load Architecture
Activity | Why It Is Commonly Misclassified as Rest | What Load It Actually Contains | Why It Can Still Trigger PEM |
Sitting on the floor painting with a child for 30 minutes | Seated, relational, not “exercise,” appears gentle | Forward posture, visual tracking, decision-making, fine motor work, emotional engagement, sustained attention | Multiple low-to-moderate burdens stack into a high total load event |
Watching C-SPAN, policy coverage, or dense news | Passive viewing, seated, no obvious movement | Analysis, memory tracking, attention maintenance, emotional processing, unpredictability of content | High cognitive burden with limited recovery opportunity |
Reading legal briefs, research papers, or complex nonfiction | Quiet, seated, appears “calm” | High task depth, precision reasoning, working memory, sustained processing | Very high sustained cognitive demand despite near-zero movement |
Scrolling social media | Casual, familiar, done while resting | Rapid switching, novelty, sensory shifts, micro-decisions, emotional micro-reactivity | Fragmented but cumulative load that can quietly push the system over threshold |
Cooking a simple meal | Routine, familiar, not formal exercise | Sequencing, timing, standing, sensory input, heat, movement, decision-making | Multi-domain burden, especially costly in an already unstable system |
Talking with family, friends, or clinicians | Social, not usually counted as exertion | Listening, memory, response generation, emotional monitoring, processing speed | Moderate to high cognitive-emotional load over time |
Eating a meal | Basic necessity, usually treated as neutral | Upright posture, chewing, swallowing, autonomic coordination, sensory input, pacing decisions | Baseline physiological cost that matters when reserve is already low |
Drinking fluids | Minimal, basic self-care | Motor coordination, swallowing, autonomic cost | Low but still non-zero burden in severe or unstable states |
Watching calm, predictable TV | Passive and low complexity | Limited narrative demand, lower novelty, lower switching | Often relatively low load and closer to actual rest |
Eyes closed, fully supported, minimal input | “Doing nothing” | Cognitive, sensory, postural, and emotional demand minimized | Closest to true rest because total system load is most suppressed |
This chart matters because it names the mistake patients live inside. Many do not crash because they ignored their illness. They crash because they were never taught how to distinguish apparent rest from actual low-load conditions. The patient who ends up in the ER after what seemed like a gentle, seated, family-centered activity is not irrational, dramatic, or mismanaging their condition. She is operating within a model that never counted the task correctly in the first place.
It is also important to state explicitly that even basic physiological functions require energy. Eating, drinking, maintaining posture, and processing sensory input all draw from the same limited adaptive reserve. In a healthy system, these costs are negligible and can be ignored. In a system operating near threshold, they are not negligible. A patient may expend a meaningful portion of their available capacity simply maintaining baseline function. This explains why some individuals feel as though they are doing almost nothing and still cannot stabilize. From a systems perspective, their baseline load may already be consuming most of what is available before additional cognitive or environmental demands are introduced.
What Has Been Missing in Clinical Advice
The limitation of current guidance is not only a lack of specificity, but a lack of structural framing. Patients are told to pace, but are not taught what the body is counting as work. Clinicians may correctly identify PEM as activity-triggered, yet still communicate in a way that equates activity with visible physical exertion. This leaves patients attempting to solve a multi-domain problem using a single-domain strategy. When this mismatch persists, the consequences extend beyond symptom management. Patients become demoralized because their efforts do not produce consistent improvement. Clinicians may interpret this as disease severity or non-responsiveness to intervention. In reality, the underlying issue is that the pacing model being applied does not match the structure of the system it is attempting to control.
Once this broader architecture is understood, the illness becomes conceptually coherent. The day that appeared restful but resulted in collapse becomes intelligible. The mismatch between low visible activity and high symptom burden becomes predictable. The plateau observed under heart rate pacing alone becomes explainable. Most importantly, the pathway to stabilization becomes actionable, because patients can begin reducing forms of load that were previously invisible.
From Stabilization to Remission
This reframing has direct implications for how remission is understood and approached. Within a partial pacing model, patients may reduce a meaningful portion of physical load while continuing to exceed threshold through cognitive, sensory, autonomic, or environmental mechanisms. In this state, the system remains unstable, and any gains are often temporary because recovery is continuously offset by unrecognized burden.
Within a multi-domain pacing model, the objective shifts. The goal is not indefinite restriction, nor the elimination of meaningful activity. The goal is to reduce total system demand sufficiently and consistently to allow recovery processes to accumulate rather than being erased. This requires identifying and reducing hidden load across domains, followed by carefully matched reintroduction of demand as capacity improves. This is where the distinction between restriction and restoration becomes critical. The purpose of identifying load correctly is not to permanently shrink the patient’s life. It is to stop the repeated cycle of overloading and partial recovery that prevents stabilization. When total demand remains below threshold for a sustained period, stability increases. As stability increases, tolerance can begin to expand. This progression represents the bridge between stabilization and remission.
The most damaging misconception in PEM-driven illness is not simply that patients are doing too much. It is that almost no one has clearly defined what “too much” actually includes. Heart rate pacing established that PEM has a boundary. A systems-based pacing model demonstrates that this boundary is multi-domain, cumulative, and delayed. Once this is understood, patient experience becomes coherent rather than contradictory. What appeared random becomes structured. What felt uncontrollable becomes manageable. What seemed fixed becomes responsive to changes in how load is defined and managed.
9. Environmental and Structural Modifiers of PEM
Post-exertional malaise (PEM) is often described as a response to exertion. This framing is incomplete. PEM is more accurately understood as a context-dependent systems event, in which internal physiologic vulnerability interacts dynamically with environmental exposures and structural constraints to determine whether a threshold breach occurs and whether recovery can proceed afterward.
In infection-associated chronic conditions such as ME/CFS and Long COVID, the underlying system is already operating in a state of reduced stability, characterized by immune dysregulation, autonomic dysfunction, neuroinflammation, endothelial impairment, and disrupted energy metabolism (Komaroff and Bateman, 2021; National Academies of Sciences, 2015; Komaroff and Lipkin, 2021). Within this terrain, the body does not respond to external inputs as a stable system would. Instead, it amplifies them, integrates them, and often fails to recover from them efficiently. This means that the same activity can produce different outcomes depending on environmental and structural context. A task that is tolerable in a low-load environment may trigger PEM in a high-load environment even when the task itself is unchanged. What appears clinically as inconsistency is, in fact, the predictable outcome of an unmeasured interaction layer. The correct framing is therefore not: PEM = exertion
but:
PEM(t) = f [Load_total(t) × Threshold Sensitivity(t) − Recovery Capacity(t)]
where Load_total includes environmental and structural inputs that are almost never captured in standard models.
Environmental Load as a Physiologic Amplifier
Environmental exposures function as continuous and cumulative load multipliers acting on an already constrained system. These include particulate matter (PM₂.₅ and PM₁₀), ozone (O₃), nitrogen dioxide (NO₂), sulfur dioxide (SO₂), volatile organic compounds (VOCs), mold spores, humidity, temperature fluctuations, and barometric instability. These exposures exert measurable biologic effects across multiple systems:
activation of inflammatory signaling pathways including IL-6, TNF-α, and NF-κB
induction of oxidative stress and mitochondrial dysfunction
impairment of endothelial function and oxygen delivery
disruption of autonomic balance and heart rate variability
activation of mast cells and neuroimmune signaling
(Pope et al., 2016; Gawda et al., 2018; WHO, 2022; Theoharides et al., 2015; Afrin et al., 2020)
These pathways are not peripheral to PEM. They overlap directly with its known pathophysiology. Impaired oxygen extraction and reduced cerebral blood flow, demonstrated in invasive cardiopulmonary exercise testing and cerebral perfusion studies, are exacerbated under inflammatory and autonomic stress conditions (Systrom et al., 2022; van Campen et al., 2020). Neuroinflammatory activation further increases cognitive fatigue and reduces processing capacity (Nakatomi et al., 2014). When these mechanisms are integrated into the load model, environmental burden becomes structurally visible:
Load_total(t) = Lp + Lc + Ls + La + Le + Lm
Where Le (environmental load) is not episodic but often continuous. This distinction matters
because continuous load raises baseline system pressure and reduces the margin available for additional activity. Rather than isolating this concept, the full structure can be seen in context:
Table: Multi-Domain Load Model of PEM (Total System Burden Framework)
Load Domain | Primary Inputs | Physiologic Mechanisms | Impact on PEM |
Physical (Lp) | Activity, exertion | Metabolic strain, anaerobic transition | Direct threshold driver |
Cognitive (Lc) | Thinking, planning | Neural energy demand, perfusion shifts | Delayed destabilization |
Sensory (Ls) | Light, sound | Central sensitization | Cumulative burden |
Autonomic (La) | Posture, stress | HR variability, vascular instability | Threshold compression |
Environmental (Le) | Pollution, mold, heat | Inflammation, oxidative stress, mast cell activation | Baseline elevation + threshold lowering |
Emotional (Lm) | Social interaction | Neuroimmune signaling | Additive load |
The key insight is that environmental load is always present but rarely counted, which creates systematic underestimation of total burden.
Indoor Environments and the Recovery Constraint
If environmental exposure determines baseline load, indoor environments determine whether recovery is biologically possible. Patients with PEM-driven illness spend a majority of time in indoor environments. When these environments contain dampness, mold contamination, poor ventilation, temperature instability, or chemical off-gassing, they introduce continuous exposure to inflammatory triggers such as spores, microbial volatile organic compounds (mVOCs), and particulate irritants. Extensive environmental health literature demonstrates that damp and mold-contaminated buildings are associated with increased respiratory illness, inflammatory activation, and neurologic symptoms (Institute of Medicine, 2004; WHO, 2009; Mendell et al., 2011). In individuals with preexisting immune and autonomic dysregulation, these exposures amplify fatigue, cognitive dysfunction, and instability. The systems consequence is direct:
Recovery(t) ≤ Ongoing Load(t)
This is the recovery constraint condition. Even when exertion is reduced, recovery cannot accumulate because the system is continuously taxed by environmental burden. This explains a clinical pattern that is frequently misinterpreted:
patients pace correctly
symptoms do not stabilize
improvement plateaus or reverses
This is not failure of pacing. It is failure of the recovery environment to support physiologic restoration.
Environmental Effects on Stability Dynamics
When environmental load is incorporated into the stability equation, its impact becomes nontrivial:
Stability(t+1) = Stability(t) + R(t) − [Load_total(t) × S(t)] − F(t+Δ)
Because Le contributes to Load_total continuously, it shifts both baseline stability and sensitivity to perturbation. This framework explains why identical pacing strategies produce different outcomes across environments. It also explains why patients frequently report improvement after environmental changes that are not traditionally considered “treatment.”
Mast Cell and Neuroimmune Threshold Sensitization
The disproportionate impact of environmental exposure in infection-associated chronic conditions is not incidental, nor is it adequately explained by generic “sensitivity.” It is better understood as the result of threshold dysregulation within immune, autonomic, and neurovascular systems, with mast cells playing a particularly important role. Mast cells function as environmental sentinels distributed throughout airway, vascular, gastrointestinal, and neurologic tissues. Under stable conditions, this surveillance role is adaptive. Under unstable conditions, particularly in patients with ME/CFS, Long COVID, dysautonomia, or mast cell disorders, activation thresholds may be lowered to the point that relatively minor exposures provoke outsized systemic responses (Theoharides et al., 2015; Valent et al., 2019; Afrin et al., 2020).
That shift in threshold has major implications for PEM. When mast cells degranulate, they release histamine, prostaglandins, leukotrienes, tryptase, cytokines, and other mediators that alter vascular tone, airway reactivity, epithelial permeability, and neurologic signaling. In practical terms, this means that an exposure that might be barely noticeable to a healthy individual can trigger tachycardia, flushing, headaches, cognitive slowing, gastrointestinal instability, airway irritation, and heightened sensory reactivity in a vulnerable patient. Those reactions are not separate from PEM. They alter the terrain in which PEM develops by increasing baseline inflammatory burden, worsening autonomic control, and reducing the system’s capacity to tolerate subsequent physical or cognitive demand.
This is why environmental load often behaves nonlinearly in these populations. The issue is not just that exposures add burden. It is that they change how the body processes burden. Within the stability model, this can be understood as an increase in threshold sensitivity, such that the same total load produces a larger destabilizing effect than it would in a less sensitized state. The Microdosing Air™ framework is particularly useful here because it reframes environmental intolerance as a threshold problem occurring within an unstable immune terrain rather than as a fixed patient trait. The Terrain Response Score (TRS) makes this explicit by modeling the interaction between terrain stability, autonomic resilience, barrier integrity, and trigger potency, showing that environmental tolerance is not static but state-dependent.
What makes this physiologically important is that it links environmental sensitivity directly back to the broader logic of PEM. The patient is not merely “reactive.” The patient is operating in a system where neuroimmune activation, autonomic instability, and impaired recovery all reinforce one another. Environmental exposures therefore become both triggers and amplifiers, capable of reducing tolerance before any visible exertion begins and of prolonging instability after the initial insult has passed.
Structural and Socioeconomic Load Multipliers
Environmental sensitivity does not occur in a vacuum, and neither does exposure. Structural and socioeconomic conditions determine not only which patients are more likely to encounter harmful environments, but also which patients have the fewest resources to reduce or escape those exposures. This is one of the clearest ways in which PEM ceases to be merely a biological event and becomes a structurally patterned one.
Substandard or aging housing, moisture intrusion, inadequate ventilation, poor insulation, lack of temperature control, occupational exposure to pollutants or irritants, inability to access air filtration, and limited financial capacity for remediation all contribute to a higher baseline burden. At the same time, delays in diagnosis, lack of knowledgeable clinicians, inadequate accommodations at work or school, and inconsistent access to supportive care increase the likelihood that patients will repeatedly exceed threshold before the underlying problem is even recognized. Jason and colleagues have long shown that case recognition, access to care, and illness interpretation are not evenly distributed across populations, while Stussman and colleagues have documented the significant diagnostic and care barriers that continue to shape the experience of people living with ME/CFS (Jason et al., 2015; Stussman et al., 2020).
These factors do not simply coexist with PEM. They multiply its consequences. A patient living in a mold-prone or poorly ventilated home, without climate control, flexible work options, transportation to knowledgeable clinicians, or financial room for remediation, is not facing the same threshold dynamics as a patient with safer housing, cleaner air, better support, and a clinician who understands PEM. The biologic condition may be similar, but the structural context is not. One patient enters each day with a lower baseline burden and more room for recovery. The other enters each day already physiologically taxed by conditions they may not be able to change.
This helps explain why PEM often appears more severe, more frequent, and more persistent in patients under sustained structural strain. Their instability is not only clinical. It is produced at the intersection of physiology and environment. A body already constrained by autonomic dysfunction, immune activation, and impaired energy metabolism is then required to function inside conditions that continuously erode whatever reserve remains. In that sense, structural disadvantage is not merely a social context around illness. It is part of the load architecture of illness itself.
Environmental Burden as Disease Infrastructure
Once this is understood, environmental exposure can no longer be treated as a secondary trigger or an inconvenient side issue. At scale, it becomes a form of disease infrastructure. Housing quality, air quality, moisture control, ventilation, exposure to mold or chemical irritants, and access to stabilizing environments directly shape how chronic illness expresses itself across populations.
This reframes mold, poor air quality, and related environmental burdens as population-scale modifiers of chronic illness severity, treatment response, healthcare utilization, and disability progression. If environmental burden suppresses recovery, lowers tolerance, and blunts treatment response, then unstable housing and exposure conditions are not merely “bad for health” in a general sense. They are active contributors to flare frequency, symptom volatility, emergency care use, and apparent treatment resistance.
A patient who seems poorly responsive to therapy may be environmentally constrained rather than biologically refractory. A community with high exposure burden may show worse average stability not because its patients are fundamentally different, but because their environments continuously load the system. Seen this way, clean air, filtration, temperature control, moisture control, remediation, and safer recovery spaces are not luxuries. They are conditions that may determine whether care can work at all.
Modeled Threshold Collapse Under Environmental Load Conditions
One poorly understood feature of ME/CFS and related conditions is that minimal activity can trigger severe PEM when baseline load is already elevated. A task such as sitting on the floor and painting with a child may appear restful, but it still includes cognitive demand, posture, sensory input, and emotional engagement. Whether that task is tolerated depends on the patient’s available threshold at that moment. The missing clinical piece is that environment changes the meaning of the same activity. In a clean, low-stimulus environment, a light task may remain tolerable. In a mold-exposed home, environmental burden is continuous, raising baseline inflammatory and autonomic strain before the task even begins. During wildfire smoke, elevated particulate matter, heat, or humidity, environmental burden may become dominant, leaving little room for even modest cognitive, postural, or sensory demand.
In practical terms, the crash may appear disconnected from the activity because the system crossed threshold earlier, then expressed the failure hours or days later. The task itself did not necessarily change. The system performing the task did. As mold, poor air quality, heat, humidity, or wildfire smoke increase environmental load, the margin between baseline function and collapse narrows. Patients are not crashing from “nothing.” They are crashing from unmeasured load inside a system whose threshold has already been compressed.
Additional Environmental Triggers That Contribute to PEM Instability
Mold exposure is one of the most clinically visible environmental triggers in ME/CFS and related infection-associated chronic conditions, but it is not unique. A growing body of environmental health and immunology research demonstrates that multiple common pollutants are capable of increasing baseline physiologic load, lowering threshold tolerance, and amplifying post-exertional
symptom instability. These exposures do not act independently of disease biology. They intersect directly with the core mechanisms already implicated in ME/CFS, including mitochondrial dysfunction, neuroinflammation, endothelial impairment, autonomic dysregulation, and immune signaling abnormalities (National Academies of Sciences, 2015; Komaroff and Lipkin, 2021; Systrom et al., 2022).
As a result, even moderate environmental exposure can function as a load multiplier, increasing the likelihood that otherwise tolerable activity will result in threshold breach and delayed PEM. Although direct longitudinal studies in ME/CFS populations remain limited, the mechanistic overlap between environmental toxicology and chronic illness biology strongly supports the role of these exposures as flare amplifiers rather than incidental irritants.
Polycyclic Aromatic Hydrocarbons (PAHs)
PAHs are produced by incomplete combustion and are common in diesel exhaust, wildfire smoke, coal emissions, and charred foods. They activate the aryl hydrocarbon receptor (AhR), disrupting immune regulation, increasing cytokine imbalance, and driving oxidative stress (Boström et al., 2002; ATSDR, 2020). In ME/CFS and related conditions, this overlaps with existing immune and metabolic dysfunction, raising baseline inflammatory load and lowering tolerance to exertion. PAHs function as background amplifiers, increasing environmental load (Le) and compressing the threshold for PEM.
Particulate Matter (PM₂.₅ and PM₁₀)
Particulate matter from traffic, industry, and wildfire smoke can penetrate deep into the lungs and enter circulation. It drives systemic inflammation, endothelial dysfunction, and autonomic disruption via pathways like NF-κB and cytokine activation (Pope et al., 2016). In ME/CFS and Long COVID, where vascular and oxygen delivery issues are already present, this increases baseline load and worsens post-exertional responses. Clinically, it reduces activity tolerance and increases risk of delayed crashes.
Nitrogen Dioxide (NO₂)
NO₂, primarily from vehicle emissions, promotes airway inflammation and impairs immune defense, particularly macrophage function (Ciencewicki and Jaspers, 2007). It also contributes to oxidative stress and autonomic imbalance, lowering resilience. Within the PEM model, NO₂ reduces threshold capacity, increasing the likelihood of delayed symptom escalation even without immediate effects.
Ground-Level Ozone (O₃)
Ozone is a strong oxidant formed from sunlight interacting with pollutants. It damages airway cells and increases systemic oxidative stress (Hollingsworth et al., 2007). Because oxidative stress and mitochondrial dysfunction are already present in ME/CFS, ozone acts as a compounding stressor, increasing fatigue, headaches, and reducing recovery efficiency.
Sulfur Dioxide (SO₂)
SO₂ from industrial and fossil fuel combustion causes bronchoconstriction and contributes to secondary particulate formation (Chen et al., 2007). Beyond respiratory effects, it adds to systemic inflammatory load, increasing fatigue and reducing exertional tolerance. It contributes to environmental load (Le) and amplifies combined pollutant exposure.
Volatile Organic Compounds (VOCs)
VOCs from traffic, building materials, and indoor sources have neurotoxic and inflammatory effects and can trigger mast cell activation (U.S. EPA, 2022).mIn ME/CFS and MCAS, they contribute to cognitive dysfunction, headaches, and fatigue. Because VOC exposure is often indoor, it can continuously impair recovery, preventing stabilization even during rest.
Why This Matters for PEM Modeling
What connects all of these exposures is not simply that they can cause symptoms. It is that they shift the system before activity even begins. They:
increase baseline inflammatory load
reduce mitochondrial efficiency
destabilize autonomic regulation
amplify neuroimmune signaling
In the language of the model:
Le ↑ → Load_total ↑ and Threshold ↓
This dual effect is what makes environmental exposure so powerful. It does not just add to load. It reduces the system’s ability to handle load. This is why a patient can move from tolerating moderate activity to crashing from minimal activity without any apparent change in behavior. The system itself has changed.
Weather and Atmospheric Volatility as Dynamic Drivers of PEM
Weather is often treated as a passive background condition in clinical reasoning, but in patients with ME/CFS and related infection-associated chronic conditions, it functions as a dynamic, continuously shifting load input that can materially alter threshold stability across hours rather than days. Unlike fixed environmental exposures such as indoor mold, atmospheric variables fluctuate in real time, introducing instability into the relationship between load and capacity even when patient behavior remains constant. This helps explain a common but poorly integrated clinical observation: patients who are stable in the morning may experience disproportionate fatigue, autonomic symptoms, or cognitive decline later in the day without any meaningful change in activity. Rather than reflecting inconsistency, this pattern reflects a time-dependent shift in environmental load interacting with an already vulnerable physiologic system. Within the total load framework, weather can be expressed as a continuous modifier of environmental burden:
Le(t) = f [PM₂.₅, ozone, humidity, temperature, barometric pressure, aeroallergens]
This formulation matters because it makes explicit that Load_total is not behaviorally bounded. Even with strict pacing, total system demand can rise due to external conditions, narrowing the margin between baseline function and threshold. This is consistent with broader environmental health literature demonstrating that short-term fluctuations in air quality, temperature, and pressure can produce measurable physiologic effects, including changes in inflammatory signaling, cardiovascular strain, and autonomic function (Pope et al., 2016; Brook et al., 2010; World Health Organization, 2021).
Barometric Pressure and Autonomic Vulnerability
Barometric pressure variation is one of the most consistently reported yet under-integrated triggers in ME/CFS and dysautonomia. Rapid drops in atmospheric pressure, typically associated with incoming storms or frontal systems, have been linked to increased reports of fatigue, headache, joint pain, and cognitive dysfunction across multiple chronic illness populations. Although the exact mechanisms remain under investigation, several physiologic pathways are plausible and supported by related research.
Changes in barometric pressure influence vascular tone, oxygen partial pressure, and autonomic regulation, all of which are already dysregulated in ME/CFS and Long COVID. Studies examining orthostatic intolerance and cerebral blood flow have demonstrated that patients with ME/CFS exhibit impaired cerebral perfusion and exaggerated hemodynamic responses to positional and physiologic stressors (van Campen et al., 2020; Rowe et al., 2014). In this context, even modest atmospheric shifts may increase the burden on compensatory autonomic mechanisms, effectively raising the cost of maintaining homeostasis. This can be modeled as an increase in autonomic load (La) alongside an increase in threshold sensitivity (S), where the same external demand produces a larger destabilizing effect. Research on weather-related pain and neurologic symptom fluctuation further supports the role of barometric pressure as a modulator of sensory and inflammatory pathways (Prince et al., 2004; Mukamal et al., 2009). For patients, the lived experience is clear: activities that were tolerable under stable conditions become destabilizing during periods of atmospheric change, even when physical exertion is unchanged.
Temperature Extremes and Thermoregulatory Demand
Temperature introduces a distinct and well-characterized physiologic burden through thermoregulatory demand, which requires coordinated cardiovascular, autonomic, and metabolic responses. In ME/CFS, where mitochondrial function, vascular regulation, and autonomic control are already impaired, these demands are amplified. Heat exposure increases cardiovascular strain through vasodilation, promotes fluid loss, and exacerbates orthostatic intolerance, all of which can reduce effective circulating volume and increase heart rate (Rowe et al., 2014; Raj et al., 2021). Epidemiologic studies have shown that heat exposure is associated with increased fatigue, cardiovascular stress, and hospital utilization, particularly in vulnerable populations (Gasparrini et al., 2015; Basu, 2009). Cold exposure, while less frequently discussed, increases metabolic demand through thermogenesis and vasoconstriction, which can further strain already compromised energy systems.
From a systems perspective, temperature contributes simultaneously to environmental load (Le) and autonomic load (La), increasing total demand while also reducing recovery efficiency. This dual effect is critical. It explains why patients often report not just discomfort, but reduced functional capacity and increased crash risk during temperature extremes, even in the absence of increased activity. The system is not simply reacting to temperature; it is reallocating limited energy toward maintaining internal stability.
Humidity, Bioaerosols, and Immune Activation
Humidity operates as a multiplier within the environmental system, influencing both biological exposure and physiologic strain. Elevated humidity promotes the growth and aerosolization of mold, dust mites, and microbial fragments, increasing exposure to bioaerosols that can activate immune and inflammatory pathways (Mendell et al., 2011; Fisk et al., 2007). In individuals with mast cell activation or heightened immune sensitivity, these exposures can increase cytokine signaling, histamine release, and autonomic instability.
At the same time, high humidity impairs evaporative cooling, increasing perceived exertion and thermoregulatory burden. Low humidity introduces a different set of challenges, including mucosal drying, increased susceptibility to particulate penetration, and airway irritation. Both extremes can destabilize the system, not through a single pathway, but through combined effects on immune activation, respiratory function, and autonomic balance. These mechanisms align with broader evidence linking environmental exposures to systemic inflammation and symptom exacerbation in chronic illness populations (Nijs et al., 2012; Afrin et al., 2020). Within the PEM framework, humidity does not act as a discrete trigger. It alters the baseline conditions under which all other loads are processed, shaping both threshold and recovery.
Wildfire Smoke and Acute Load Amplification
Wildfire smoke represents one of the clearest real-world demonstrations of how environmental conditions can rapidly and profoundly alter system stability. Smoke exposure introduces a complex mixture of fine particulate matter (PM₂.₅), polycyclic aromatic hydrocarbons, carbon monoxide, and reactive gases, all of which contribute to oxidative stress, endothelial dysfunction, and systemic inflammation (Reid et al., 2016; Cascio, 2018). Short-term increases in PM₂.₅ have been associated with increased cardiovascular events, respiratory symptoms, and inflammatory markers, even in otherwise healthy populations (Pope et al., 2016; Brook et al., 2010). In patients with ME/CFS or Long COVID, where these pathways are already dysregulated, the impact is amplified. Environmental load can increase sharply:
Le ↑↑ → Load_total ↑↑ → Threshold margin ↓
This creates a condition in which baseline load approaches or exceeds threshold, leaving minimal capacity for additional demand. Patients often report rapid symptom escalation, including fatigue, cognitive dysfunction, headaches, and autonomic instability, during wildfire events. Importantly, these changes can occur without any increase in activity, demonstrating that environment alone can drive threshold breach.
Weather as a Threshold Compression System
When considered together, atmospheric variables function not simply as additive burdens but as a threshold compression system. They increase baseline load, increase autonomic demand, amplify inflammatory signaling, and reduce recovery efficiency, all of which narrow the margin between baseline function and collapse.
This relationship can be expressed as:
Effective Threshold(t) = Base Threshold − Weather Impact(t)
At the same time, recovery dynamics are altered:
R(t) ↓ as environmental stress ↑
This dual effect is critical. It explains why patients may experience worsening symptoms even when they reduce activity. The system is not only taking on more load; it is also recovering less effectively from that load. This aligns with emerging models of chronic illness that emphasize impaired homeostasis and reduced physiologic resilience as central features of disease (Komaroff and Lipkin, 2021; Systrom et al., 2022).
Clinical and Modeling Implications
Incorporating weather into PEM modeling resolves several persistent inconsistencies in both patient experience and clinical interpretation. It explains why symptoms fluctuate without clear behavioral triggers, why pacing strategies may appear to fail under certain conditions, and why recovery is inconsistent even when activity is carefully controlled. It also provides a mechanistic basis for widely reported sensitivities to storms, heat, humidity, and air quality changes.
More importantly, it expands the scope of meaningful intervention. Environmental awareness, atmospheric monitoring, and adaptive pacing based on real-time conditions become central components of disease management rather than peripheral considerations. This aligns with broader trends in digital health, where environmental and physiologic data are increasingly integrated to predict and prevent adverse outcomes (Topol, 2019). Within CYNAERA’s framework, systems such as VitalGuard™ operationalize this logic by integrating atmospheric data with individualized sensitivity profiles to model flare risk dynamically. By linking external variability to internal system response, this approach reframes weather not as an uncontrollable external factor, but as a forecastable and actionable component of threshold management.
Synthesis: From Environmental Exposure to Threshold Behavior
Taken together, atmospheric variables do not function as isolated triggers but as dynamic modifiers of system stability, continuously shaping both baseline load and recovery capacity. This reframes weather from an external nuisance into a central component of PEM architecture. The key shift is conceptual but clinically decisive: patients are not simply reacting to activity, they are responding to activity within a moving environmental system. In this model, threshold is not fixed across the day. It is continuously recalibrated by atmospheric conditions that alter vascular tone, autonomic demand, inflammatory signaling, and oxidative stress. As a result, the same task can exist in three entirely different physiologic contexts within a single 24-hour period. When weather volatility is ignored, this produces the appearance of randomness. When it is included, the pattern becomes coherent. This is most clearly illustrated at the level of a single patient day.
Modeled Patient Timeline: Stability to Crash Under Weather Shift
A patient begins the day in a relatively stable indoor environment. Overnight recovery has partially restored capacity, air quality is acceptable, and barometric pressure is steady. At this point:
baseline load is moderate
threshold margin is positive
light cognitive or physical activity remains tolerable
The patient engages in routine morning activity, such as light conversation, preparing food, or working briefly on a task. Under these conditions, the system remains below threshold:
Load_total < Threshold → stability maintained
By late morning, atmospheric conditions begin to shift. A weather front moves in, producing a drop in barometric pressure. At the same time, regional air quality deteriorates due to increased particulate matter, either from urban pollution accumulation or wildfire drift. These changes are not perceived as discrete events by the patient, but they alter system dynamics in measurable ways:
autonomic demand increases as vascular regulation becomes less efficient
inflammatory signaling rises in response to particulate exposure
oxidative stress increases, reducing mitochondrial efficiency
effective threshold begins to narrow
At this stage, the patient may still feel functional, but the system has already changed:
Le(t) ↑ → Load_total ↑ and Effective Threshold ↓
In the early afternoon, the patient performs what would normally be considered a low-intensity activity, such as sitting upright and engaging in a cognitively light task. Under morning conditions, this activity would remain well within capacity. Under current conditions, however, the system is operating with reduced margin.
The same activity now produces:
increased cognitive demand relative to available energy
increased autonomic strain due to pressure instability
additive inflammatory burden from environmental exposure
This pushes the system toward threshold:
Load_total ≈ Threshold
Importantly, the patient may not feel immediate distress. This is where clinical misinterpretation often occurs. Because the activity feels manageable in the moment, it is not recognized as a trigger. However, the system has already crossed into a state of subclinical threshold breach, where recovery demand exceeds recovery capacity. By late afternoon or evening, delayed effects begin to emerge:
fatigue intensifies disproportionately
cognitive dysfunction becomes more pronounced
autonomic symptoms increase
recovery does not consolidate
This is the delayed flare expression:
F(t+Δ) = accumulated load + reduced recovery efficiency
The patient experiences this as a crash that appears disconnected from the activity that preceded it. In reality, the crash reflects the interaction between environmental shift and task load within a compressed threshold window.
Closing Integration
This timeline illustrates a core principle that has been repeatedly observed but rarely formalized: PEM is not solely triggered by what a patient does, but by what the system is capable of absorbing at that moment in time. Weather and atmospheric variability directly influence that capacity.
By integrating these variables into the load model, the apparent unpredictability of PEM begins to resolve into a structured, interpretable pattern. Threshold breaches that once seemed random can be understood as the result of stacked load interacting with dynamic environmental constraints. This not only improves clinical interpretation but also opens the door to more effective intervention, where pacing is adjusted not just to activity, but to the conditions under which that activity occurs. In this context, atmospheric awareness becomes more than observational. It becomes a practical tool for preserving stability, preventing threshold breaches, and reducing the frequency and severity of post-exertional decline.
10. PEM and the Path to Remission
If post-exertional malaise is understood as a threshold-dependent systems failure, then meaningful recovery cannot be defined solely by symptom reduction in isolation. A patient may report lower fatigue, fewer headaches, or better daily function at a given moment while still operating within a highly unstable system that remains unable to tolerate ordinary stress without disproportionate collapse. In this context, improvement must be evaluated not only through symptom change, but through changes in system stability, durability, flare behavior, functional capacity, and resilience under real-world conditions. That distinction matters because it separates temporary relief from meaningful recovery, and apparent improvement from true remission.
This is precisely the problem the CYNAERA Remission Standard™ was designed to solve. Across chronic illness, autoimmune disease, dysautonomia, oncology, and post-viral conditions, remission is often inferred from symptom reduction, biomarker normalization, or treatment response at a single point in time. Yet these definitions repeatedly fail in conditions characterized by delayed collapse, flare dynamics, environmental sensitivity, and relapsing-remitting behavior. A patient may look improved in clinic while remaining fundamentally unstable in life. A system may appear calm in protected conditions while still lacking the resilience required to hold gains under cognitive, physical, autonomic, or environmental demand. The CYNAERA Remission Standard™ corrects for that by defining remission as a measurable state of sustained stability, durability, functional capacity, flare control, and resilience under real-world conditions.
In PEM-driven illness, this is especially important because PEM is not simply one symptom among many. It is the clearest visible signal of underlying systems instability under load. It reveals whether the system can tolerate stress, how it fails when it cannot, and how effectively it recovers afterward. For that reason, remission cannot be meaningfully assessed without looking directly at how PEM changes over time. A system moving toward remission should demonstrate not only lower symptom intensity, but higher exertional thresholds, fewer and less severe crashes, shorter recovery windows, less delayed amplification after stress, and improved tolerance across physical, cognitive, sensory, orthostatic, emotional, and environmental domains (Komaroff and Bateman, 2021; Wirth and Scheibenbogen, 2021; Davenport et al., 2019; Jason et al., 2009). Rather than disappearing abruptly, PEM should become less frequent, less destabilizing, and less central to overall system behavior.
CYNAERA Remission Standard™ as the Outcome Layer
The CYNAERA Remission Standard™ defines remission as a state-dependent outcome measured across five domains: stability, durability, function, flare control, and resilience. This prevents overcalling remission based on partial gains, distinguishing between visible improvement and true system-level stability over time.
UNCT and Biological Logic
Under UNCT, PEM reflects instability across immune, metabolic, autonomic, and neuroinflammatory systems. Remission represents partial restoration of coordination across these domains, raising threshold, reducing flare penalty, and improving recovery capacity. It is not full normalization, but sufficient coherence to tolerate load without collapse.
SymCas™ and Time-Dependent PEM
SymCas™ models PEM as a delayed cascade rather than a point event. Remission progression is reflected in narrower delayed responses, reduced severity, and faster recovery, making PEM one of the most sensitive dynamic outcome measures.
BLI™ and GCLR™: Multi-Domain Load Matching
Remission requires matching total system load, not just physical exertion. BLI™ estimates capacity, while GCLR™ captures cognitive, sensory, and task complexity.
Risk = GCLR_eff − BLI + Environmental Penalty
As remission develops, capacity stabilizes and expands, allowing higher demand without delayed collapse.
VitalGuard™ and Environmental Readiness
Environmental load directly shapes threshold and recovery. Reductions in particulate matter, humidity, mold, and other exposures lower baseline burden and expand threshold margin:
Le ↓ → Load_total ↓ → Threshold margin ↑ → Stability ↑
This explains why patients plateau despite appropriate pacing when environmental burden remains high.
STAIR and Sequencing
Recovery is staged. Interventions must align with system readiness, as mistimed escalation can worsen instability even when the intervention itself is appropriate.
CDF™ and Subtype Variation
Remission pathways differ by dominant drivers such as autonomic dysfunction, immune volatility, or environmental sensitivity. CDF™ enables subtype-specific targeting.
Remission Pathways™ and CRISPR²™
Remission Pathways™ guide progression from instability to durability, while CRISPR²™ integrates timing, stabilization, and personalized recovery to improve long-term outcomes.
PEM as the Core Remission Signal
PEM remains the most meaningful indicator of remission because it reflects system behavior under load. Improvements are seen as reduced volatility, fewer crashes, improved recovery, and greater tolerance to real-world stress.
Closing Integration
Remission is not symptom suppression but restoration of system stability under real-world conditions. Within the CYNAERA architecture, SymCas™, BLI™, GCLR™, VitalGuard™, STAIR, and CRISPR²™ integrate timing, load, and environment into a unified model where PEM defines readiness, guides intervention, and measures outcome.
AIM Integration and Applied PEM Intelligence
This paper is designed to function as more than a standalone publication. Within CYNAERA, it operates as a canonical reference layer through the Aligned Intelligence Method (AIM™), a human-readable, machine-interpretable knowledge framework that embeds interpretive guardrails directly into source documents . Tools such as IACC Twin™, and The Eve Research Projects GPT, can interpret symptom patterns using a shared definition of PEM as a threshold-dependent, multi-domain system failure.
11. Conclusion
Post-exertional malaise has long been recognized as the defining feature of ME/CFS, yet it has remained conceptually under-integrated within the clinical, research, and policy frameworks that shape how the condition is understood and managed. The result has been a persistent and consequential disconnect between patient experience and medical interpretation, one that has contributed to delayed diagnosis, inappropriate treatment strategies, inconsistent research findings, and systemic underestimation of disease burden. This paper has argued that PEM should not be treated as a secondary or descriptive symptom, but as the primary observable signal of systems-level instability. Its defining characteristics, delayed onset, disproportionate response to stress, multisystem involvement, and prolonged recovery, reflect a failure of coordinated adaptation across immune, metabolic, autonomic, neuroinflammatory, and environmental-response systems. These features place PEM outside the scope of traditional models that rely on immediate, linear cause-and-effect relationships or static measurement approaches.
The failure to account for timing, threshold dynamics, and multi-domain load interaction has led to widespread misclassification of patients, underrecognition of severity, and the repeated misinterpretation of both clinical and trial outcomes. Frameworks that rely on single timepoint assessments or narrow definitions of exertion remain structurally misaligned with a condition defined by variability, delayed response, and cumulative burden. As a result, meaningful signals of deterioration or improvement are often missed, and treatment effects are frequently misunderstood.
By reframing PEM as a threshold-dependent systems failure, a more coherent and clinically actionable model emerges. In this model, exertion is no longer limited to physical activity but is understood as one component of total system load, alongside cognitive, sensory, autonomic, environmental, and emotional demand. Symptom exacerbation occurs when cumulative load exceeds a reduced and dynamic capacity for adaptation. This framework accounts for delayed onset, variability between patients, and the cumulative impact of repeated stress without adequate recovery.
Importantly, this reframing does not only clarify the problem. It defines the pathway forward.
A PEM-centered model enables longitudinal measurement, allowing clinicians and researchers to observe how the system behaves over time rather than relying on isolated snapshots. It supports precision stratification, recognizing that patients differ in their dominant drivers of instability and therefore require different stabilization strategies. It enables state-dependent intervention design, where timing, readiness, and environmental context are treated as core variables rather than afterthoughts. Most critically, it provides a foundation for measuring meaningful recovery.
Within the CYNAERA architecture, this progression becomes structured and measurable. UNCT defines the underlying collapse dynamics. SymCas™ captures the temporal cascade of delayed response. BLI™ and GCLR™ quantify multi-domain load and capacity mismatch. VitalGuard™ integrates environmental burden into threshold modeling. STAIR governs stabilization sequencing. CDF™ enables subtype-specific interpretation. Remission Pathways™ and CRISPR²™ guide progression from instability to durability.
The CYNAERA Remission Standard™ then functions as the outcome layer, defining remission as a state of sustained stability, durability, functional capacity, flare control, and resilience under real-world conditions. This integration resolves a longstanding problem in chronic illness research and care. It replaces fragmented, domain-specific interpretations with a unified systems model that aligns biological behavior, patient experience, and measurable outcomes. It also reframes remission itself, not as the absence of symptoms at a single point in time, but as the restoration of adaptive capacity across time and context.
The central challenge has never been that PEM is unknowable or inherently inconsistent. It is that it has been studied using tools and assumptions that do not match its structure. Correcting that mismatch is essential. Until PEM is modeled as a dynamic, threshold-based, multi-domain system, progress will remain limited, regardless of advances in individual areas of research. Conversely, once PEM is understood and measured on its own terms, it becomes one of the most powerful signals available in medicine. It reveals not only how the system fails, but how it recovers. It provides a direct window into stability, resilience, and readiness for intervention. And it offers a scalable, cross-condition framework for understanding remission in complex chronic illness.
Ultimately, the goal is not simply to reduce symptoms. It is to restore the system’s ability to engage with life without triggering collapse. In PEM-driven illness, that is the difference between temporary improvement and durable recovery. And that distinction is where meaningful progress begins.
Special Author's Note
This work represents a first-of-its-kind effort to operationalize post-exertional malaise (PEM) as a computable systems variable. Rather than treating PEM as a passive outcome of exertion, this framework models it as an active, time-dependent signal that governs system stability, predicts flare trajectories, and defines the boundaries of safe intervention and remission timing.
FAQ: Post-Exertional Malaise in ME/CFS
What is post-exertional malaise (PEM)?
Post-exertional malaise is a delayed, disproportionate worsening of symptoms following physical, cognitive, sensory, autonomic, or environmental stress. It reflects a failure of coordinated physiological adaptation across multiple systems, including immune, metabolic, autonomic, and neurocognitive domains, rather than simple fatigue or energy depletion.
How is PEM different from ordinary fatigue or burnout?
Ordinary fatigue occurs during or immediately after activity and typically resolves with rest. PEM is characterized by delayed onset, often 12 to 72 hours after exertion, multisystem involvement, and prolonged recovery. It represents a pathological response to stress rather than a normal physiologic limit being reached.
Why does PEM occur hours or days after exertion?
The delayed onset reflects time-dependent dysfunction in regulatory systems. Initial compensation may mask early instability, but downstream processes such as immune signaling, metabolic stress, impaired perfusion, and autonomic dysregulation amplify over time, producing a cascade effect that becomes clinically visible after a lag period.
Can non-physical activities trigger PEM?
Yes. Cognitive load, sensory input, emotional stress, orthostatic strain, and environmental exposures all contribute to total system load. Activities such as sustained concentration, complex decision-making, conversations, or exposure to heat, noise, or poor air quality can independently or cumulatively trigger PEM.
Why do some patients experience more severe PEM than others?
Severity is determined by underlying system stability and threshold sensitivity. Factors such as immune dysregulation, autonomic instability, mitochondrial dysfunction, environmental burden, and illness duration influence how quickly threshold is exceeded and how severe and prolonged the resulting crash becomes.
Does PEM mean patients are deconditioned?
No. Evidence shows that PEM is associated with abnormalities in metabolic function, oxygen utilization, and autonomic regulation that occur independently of deconditioning. In many cases, these abnormalities precede reductions in activity and cannot be explained by inactivity alone.
How should PEM be measured in clinical care or research?
PEM requires longitudinal, trajectory-based assessment rather than single timepoint evaluation. Clinically meaningful measurement includes tracking delayed symptom response following defined stressors, flare frequency and severity, recovery duration, and variability over time. Static symptom scales alone are insufficient.
What does improvement in PEM look like?
Improvement is typically gradual and reflects changes in system behavior rather than immediate symptom resolution. Key indicators include increased tolerance to multi-domain load, reduced frequency and severity of crashes, shorter recovery windows, and less pronounced delayed amplification following stress.
Why is PEM critical for clinical trials?
PEM directly affects treatment response and outcome interpretation. Trials that do not account for delayed effects, baseline instability, and variability risk misclassifying responders and missing true therapeutic signal. PEM-aware design improves endpoint accuracy and reduces false conclusions.
Is PEM unique to ME/CFS?
PEM is most strongly associated with ME/CFS but is also observed in related conditions, particularly Long COVID and other infection-associated chronic conditions. Its presence is a key differentiator from other causes of fatigue and is central to accurate diagnosis and classification
CYNAERA Framework Papers
This paper draws on a defined subset of CYNAERA Institute white papers that establish the methodological and analytical foundations of CYNAERA’s frameworks. These publications provide deeper context on prevalence reconstruction, remission, combination therapies and biomarker approaches. Our Long COVID Library, ME/CFS Library, Lyme Library, Autoimmune Library and CRISPR Remission Library are also in depth resources.
Author’s Note:
All insights, frameworks, and recommendations in this written material reflect the author's independent analysis and synthesis. References to researchers, clinicians, and advocacy organizations acknowledge their contributions to the field but do not imply endorsement of the specific frameworks, conclusions, or policy models proposed herein. This information is not medical guidance.
Patent-Pending Systems
Bioadaptive Systems Therapeutics™ (BST) and affiliated CYNAERA frameworks are protected under U.S. Provisional Patent Application No. 63/909,951. CYNAERA is built as modular intelligence infrastructure designed for licensing, integration, and strategic deployment across health, research, public sector, and enterprise environments.
Licensing and Integration
CYNAERA supports licensing of individual modules, bundled systems, and broader architecture layers. Current applications include research modernization, trial stabilization, diagnostic innovation, environmental forecasting, and population level modeling for complex chronic conditions. Basic licensing is available through CYNAERA Market, with additional pathways for pilot programs, institutional partnerships, and enterprise integration.
About the Author
Cynthia Adinig is the founder of CYNAERA, a modular intelligence infrastructure company that transforms fragmented real world data into predictive insight across healthcare, climate, and public sector risk environments. Her work sits at the intersection of AI infrastructure, federal policy, and complex health system modeling, with a focus on helping institutions detect hidden costs, anticipate service demand, and strengthen planning in high uncertainty environments.
Cynthia has contributed to federal health and data modernization efforts spanning HHS, NIH, CDC, FDA, AHRQ, and NASEM, and has worked with congressional offices including Senator Tim Kaine, Senator Ed Markey, Representative Don Beyer, and Representative Jack Bergman on legislative initiatives related to chronic illness surveillance, healthcare access, and data infrastructure. In 2025, she was appointed to advise the U.S. Department of Health and Human Services and has testified before Congress on healthcare data gaps and system level risk.
She is a PCORI Merit Reviewer, currently advises Selin Lab at UMass Chan, and has co-authored research with Harlan Krumholz, MD, Akiko Iwasaki, PhD, and David Putrino, PhD, including through Yale’s LISTEN Study. She also advised Amy Proal, PhD’s research group at Mount Sinai through its CoRE advisory board and has worked with Dr. Peter Rowe of Johns Hopkins on national education and outreach focused on post-viral and autonomic illness. Her CRISPR Remission™ abstract was presented at CRISPRMED26 and she has authored a Milken Institute essay on artificial intelligence and healthcare.
Cynthia has been covered by outlets including TIME, Bloomberg, Fortune, and USA Today for her policy, advocacy, and public health work. Her perspective on complex chronic conditions is also informed by lived experience, which sharpened her commitment to reforming how chronic illness is understood, studied, and treated. She also advocates for domestic violence prevention and patient safety, bringing a trauma informed lens to her research, systems design, and policy work. Based in Northern Virginia, she brings more than a decade of experience in strategy, narrative design, and systems thinking to the development of cross sector intelligence infrastructure designed to reduce uncertainty, improve resilience, and support institutional decision making at scale.
Key References
Carruthers, B.M. et al. (2011) ‘Myalgic encephalomyelitis: International Consensus Criteria’, Journal of Internal Medicine, 270(4), pp. 327–338.
Chu, L. et al. (2020) ‘Severity of myalgic encephalomyelitis/chronic fatigue syndrome symptoms and their association with post-exertional malaise’, Journal of Translational Medicine, 18, p. 101.
Cook, D.B. et al. (2017) ‘Neural consequences of post-exertion malaise in myalgic encephalomyelitis/chronic fatigue syndrome’, Brain, Behavior, and Immunity, 62, pp. 87–99.
Davenport, T.E. et al. (2019) ‘Conceptual model for physical therapist management of myalgic encephalomyelitis/chronic fatigue syndrome’, Physical Therapy, 99(6), pp. 757–768.
Davis, H.E. et al. (2023) ‘Long COVID: major findings, mechanisms and recommendations’, Nature Reviews Microbiology, 21(3), pp. 133–146.
Fluge, Ø. et al. (2016) ‘Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome’, JCI Insight, 1(21), e89376.
Hornig, M. et al. (2015) ‘Distinct plasma immune signatures in ME/CFS are present early in the course of illness’, Science Advances, 1(1), e1400121.
Institute of Medicine (2015) Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness. Washington, DC: National Academies Press.
Jason, L.A. et al. (2009) ‘The energy envelope theory and myalgic encephalomyelitis/chronic fatigue syndrome’, AAOHN Journal, 57(4), pp. 175–181.
Komaroff, A.L. (2019) ‘Advances in understanding the pathophysiology of chronic fatigue syndrome’, JAMA, 322(6), pp. 499–500.
Komaroff, A.L. and Bateman, L. (2021) ‘Will COVID-19 lead to myalgic encephalomyelitis/chronic fatigue syndrome?’, Frontiers in Medicine, 7, 606824.
Komaroff, A.L. and Lipkin, W.I. (2021) ‘Insights from myalgic encephalomyelitis/chronic fatigue syndrome may help unravel the pathogenesis of postacute COVID-19 syndrome’, Trends in Molecular Medicine, 27(9), pp. 895–906.
Nakatomi, Y. et al. (2014) ‘Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis’, Journal of Nuclear Medicine, 55(6), pp. 945–950.
National Institute for Health and Care Excellence (NICE) (2021) Myalgic encephalomyelitis/chronic fatigue syndrome: diagnosis and management (NG206). London: NICE.
Pope, C.A. et al. (2016) ‘Health effects of fine particulate air pollution: lines that connect’, Journal of the Air & Waste Management Association, 66(8), pp. 709–742.
Raj, S.R. et al. (2020) ‘Postural tachycardia syndrome (POTS): state of the science’, Circulation, 141(12), pp. e125–e127.
Rowe, P.C. et al. (2014) ‘Neuromuscular strain increases symptom intensity in chronic fatigue syndrome’, Pediatrics, 133(4), pp. e1105–e1112.
Snell, C.R. et al. (2013) ‘Discriminative validity of metabolic and workload measurements for identifying people with chronic fatigue syndrome’, Physical Therapy, 93(11), pp. 1484–1492.
Systrom, D.M. et al. (2021) ‘Impaired systemic oxygen extraction in myalgic encephalomyelitis/chronic fatigue syndrome’, Chest, 160(2), pp. 642–651.
Tomas, C. et al. (2017) ‘Cellular bioenergetics is impaired in patients with chronic fatigue syndrome’, PLoS ONE, 12(10), e0186802.
van Campen, C.M.C., Rowe, P.C. and Visser, F.C. (2020) ‘Orthostatic symptoms and reductions in cerebral blood flow in chronic fatigue syndrome’, Clinical Neurophysiology Practice, 5, pp. 50–58.
VanNess, J.M. et al. (2010) ‘Postexertional malaise in women with chronic fatigue syndrome’, Journal of Women’s Health, 19(2), pp. 239–244.
Wirth, K. and Scheibenbogen, C. (2021) ‘A unifying hypothesis of the pathophysiology of myalgic encephalomyelitis/chronic fatigue syndrome’, Frontiers in Neurology, 12, 656229.
World Health Organization (WHO) (2022) WHO Global Air Quality Guidelines. Geneva: World Health Organization.
How to Cite this Paper
Adinig, C. (2026). Post-Exertional Malaise (PEM) and Remission: A Flare-Aware State-Dependent Systems Model for ME/CFS and Long COVID. CYNAERA. Available at: https://www.cynaera.com/post/pem-and-remission



Comments